{kind=link}
I thank Andrés Aravena for his great help and encouragement.
- Summary
- Özet
-
- Metagenomics
- 1.1. Use Of Metagenomics
- 1.2. NGS In Metagenomics
- 1.3. Problems In Metagenomics
-
- Assigning IDs To Metagenome
- 2.1. Assessing Taxonomic Composition
- 2.2. Problems In Identifying The Taxonomic Identity
- 2.3. Importance Of Assessing Taxonomy
- 2.2.1. Sequence Mapping
- 2.2.2. Binning Based On Nucleotide Composition
- 2.2.3. Metabarcoding
- 2.4. Selecting Classifiers
-
- Taxonomic Classification with Kraken
- 3.1. Simulating Data
- 3.2. Prosseccing Of Datasets
- 3.3. Results And Disscussion
-
- Further Analysis
- Supplementary
- References
- CV
Metagenomics is the study of complex and diverse communities of microbial populations without the need of isolation. The term was first to put in a view of exploring uculturable soil microorganisms in 1998 [1]. Approach of metagenomics make a dissimilar attempt from classical culture-dependent microbial studies attempt to assign properties such as metabolic activities and interactions of the entire community [2]. Metagenomics generally incorporate extraction of nucleic and amino acids from enviromental samples, sequencing the extract and multiple following analysis of output data with springing bioinformatics tools. This broad possibility of options faciliates studies, besides complicates the selection of the right tool for desired metagenomics analysis. Theoretically, metagenomics can unveil almost all properties of large group of microorganisms that substracted from soil and aqua enviroments to human gut microbiota. Theoretically, metagenomics can unveil almost all properties of large group of microorganisms that cannot isolated in laboratory [7].
Rapid development of Next-Generation Sequencing (NGS) revolutionized metagenomics projects. 454 pyrosequencer was the first widely used sequncing technology. Although it was used in many studies in the early years, datasets also can be found generated via this technology. Great coverage produced by Illumina and its low cost per Mb have made Illumina largely appealing high throuput technology in metagenomics studies at present [3]. Long-read sequencing technologies can allow for precise taxonomic classification and funtional annotation, but the cost of the technologies, high error rates and the excessive need of input DNA requirements restrict their use.
For examining the microbial organisms, pure-culture technics were the gold standart for laboratory experiments. Single line of the organism ,independent from their neighborhood, is the only desired thing to be grown on dishes. Altough many microbial organisms coexist in complex communities in which they interact in complex ways [9]. Culture dependant methods are inadequate in revealing these complex interactions by growing only one constituent of community. Eventual intent of microbial genomics would sequence all species if they were culturable in laboratory conditions. But only few species among the microbial organisms are suitable for culturing [8] and few have sequence of complete genome. It appears to be that about one percent of bacteria solely can be cultured in laboratory. The growth of the bacteria is due to the fact that they are effective in adapting to the laboratory conditions rather than the impact on the community.
One important task of metagenomics studies is that determine taxonomic composition of all organisms in enviromental sample as it is valuable to find their disttribution in community [10]. The questions "Who is there?" and "What are they doing?" are connected as inhabitants of the community equally take part in ecosystem functions. Therefore, it provides great knowledge about composition of the community and give an outline of functions and interactions of the community.
Genome sequences that we have today is far from covering all living organisms [11], so that assigning sequences that encountered first time may skipped over due to lack of complete genome sequences. Likewise, ranks in microbial organisms are not approved generally, the degree of a new taxon that appears unlike the known place, thus, is diffucult to decide [12]. Lastly, The horizontal gene transfer between microbial organisms which notable in prokaryotic world, discrepancies between the sequence phylogeny and the organism phylogeny [13], leading to load additional analyses.
Results of this analyses is crucial because certain phylogenetic groupings can be associated with important functions. The diversity of a microbial community is considered to be an indication of the system's durability such as the ability to maintain its functioning when conditions change [14].
Generally three aproaches of taxonomic classification or alias taxonomic binning can be included in groups: mapping of sequences to a database of reference genomes, binning of sequences based on their composition of nucleotide k-mers and classification of reads deriving from certain barcoding regions, particularly the small subunit (SSU) rRNA genes [15].
Relatively simple task of mapping reads or contigs to genome is that all reads from metagenome are coupled to a list of reference genomes such as found in NCBI Refseq [16], GOLD [18], KEGG [19] or Human Microbiome Project [17] databases, utilizing sequence similarity. Tools like BWA [8], Bowtie2 [9] or Diamond [] are used to map, only allowing slight range of dissimilarity.
Kraken [5] provides fast identification of all reads in sample to the database. It achieve this by matching exact k-mers instead of alignment of reads that requires lot more computational power.
Simulating metagenomic data is utilized for testing new tools and developing new methods. The realistic metagenomic reads that displayed in this paper were generated by InSilicoSeq [4]. InSilicoSeq is a software that uses multiple genomes to generate real-like Illumina reads. We downloaded Genomes from Ncbi Genbank.
#git clone https://github.com/mw55309/Kraken_db_install_scripts.git
perl $(download_bacteria.pl, download_archaea.pl, download_fungi.pl, download_protozoa.pl, download_viral.pl)
library(readr)
set.seed(234)
domains <- c("archaea", "bacteria", "fungi", "protozoa", "viral")
read_assembly <- function(domain) {
# read all sequences descriptions
filename <- paste0(domain,"_assembly_summary.txt")
assembly <- read_delim(filename, "\t", escape_double = FALSE, trim_ws = TRUE, skip = 1)
colnames(assembly)[1] <- "assembly_accession"
# filter and keep only Complete Genome
genomes <- assembly[assembly$assembly_level=="Complete Genome",]
return(genomes)
}
genomes <- lapply(domains, read_assembly)
names(genomes) <- domains
rand_files_domain <- function(domain, N) {
# select N species_taxid randomly
species <- unique(genomes[[domain]]$species_taxid)
selected_species_id <- sample(species, N)
# select randomly one strain for each species
get_names <- function(species_id) {
selected_species <- subset(genomes[[domain]], species_taxid==species_id)
selected_seq_idx <- sample.int(nrow(selected_species), 1)
selected_sequences <- selected_species[selected_seq_idx, ]
paste(selected_sequences$assembly_accession, selected_sequences$asm_name, "genomic.tax.fna", sep="_")
}
seq_filenames <- sapply(selected_species_id, get_names)
return(paste(domain, seq_filenames, sep="/"))
}
archaea <- rand_files_domain("archaea", 100)
bacteria <- rand_files_domain("bacteria", 2000)
fungi <- rand_files_domain("fungi", 5)
protozoa <- rand_files_domain("protozoa", 1)
viral <- rand_files_domain("viral", 4000)
get_files_domain <- function(domain, N, veto) {
# get all filenames in domain
seq_filenames <- paste(genomes[[domain]]$assembly_accession, genomes[[domain]]$asm_name, "genomic.tax.fna", sep="_")
seq_filenames <- paste(domain, seq_filenames, sep="/")
return(sample(setdiff(seq_filenames, veto), N))
}
test_archaea <- get_files_domain("archaea", 100, archaea)
test_bacteria <- get_files_domain("bacteria", 1500, bacteria)
test_fungi <- get_files_domain("fungi", 5, fungi)
test_protozoa <- get_files_domain("protozoa", 1, protozoa)
test_viral <- get_files_domain("viral", 3000, viral)
#install
sudo -H pip3 install insilicoseq
#simulation
awk '{gsub(" ","_"); print "/home/db/Kraken/seq/"$0}' /home/db/Kraken/db/test_fname.txt | xargs iss generate --model miseq --output miseq --abundance uniform --genomes
#download taxonomy
kraken-build --download-taxonomy --db k15min7
#add all fasta files
for i in $(cat train_fname.txt); do kraken-build --add-to-library /home/db/Kraken/seq/$i --db k15min7; done
#build database
kraken-build --build --db k15min7
#run kraken
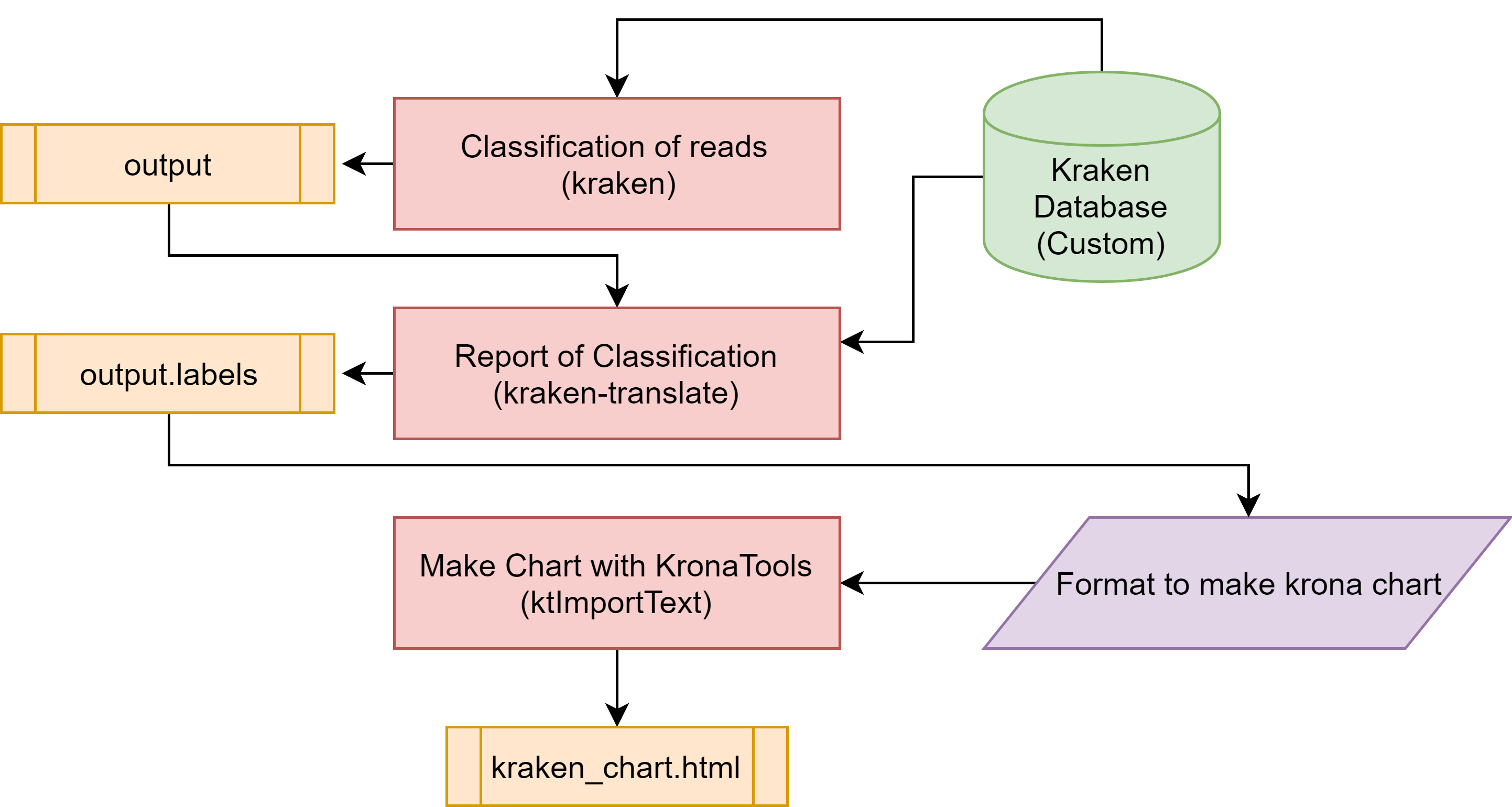
kraken --db k15min7 --threads 22 --output output.kraken --fastq-input --paired /home/db/Metagenomics/InSilicoSeq/2019-04-17/miseq_R*
#report
kraken-report --db k15min7 output.kraken > output.kraken.report
#put labels
kraken-translate --db k15min7 output.kraken > output.kraken.labels
#code for true-false table
from ete3 import NCBITaxa
import sys
ncbi = NCBITaxa()
wanted = [u'phylum', u'class', u'order', u'family', u'genus', u'species']
def get_lineage(txid):
lineage = ncbi.get_lineage(txid)
rnk = ncbi.get_rank(lineage)
return(dict([(rnk[x],x) for x in lineage if rnk[x] in wanted]))
def compare_lineage(a,b):
if a is None:
return ("NA")
elif b is None:
return ("NA")
elif a==b:
return("1")
else:
return("0")
for line in sys.stdin:
if line[0]!='C':
continue
cols = line.strip().split()
original_txid = cols[1].split("|")[2].split("_")[0]
assigned_txid = cols[2]
try:
o_lineage = get_lineage(original_txid)
a_lineage = get_lineage(assigned_txid)
comp = [compare_lineage(o_lineage.get(rank, None), a_lineage.get(rank, None))
for rank in wanted]
print(original_txid, assigned_txid, " ".join(comp))
except:
pass
#libraries
library(ggplot2)
library(data.table)
#summary function
read.kraken <- function(fname) {
x <- read.table(fname, na.strings = "NA",
col.names = c("or_taxid", "as_taxid", "ph", "cl", "or", "fm", "ge", "sp"))
# bacstat <- apply(bac, 2, sum, na.rm = TRUE)
x <- x[, -(1:2)]
ones <- colSums(x, na.rm = TRUE)
noes <- colSums(is.na(x))
zeros <- nrow(x) - ones - noes
return(data.frame(Yes=ones, Maybe=noes, No=zeros,
rank=factor(c("ph", "cl", "or", "fm", "ge", "sp"), ordered = FALSE)))
}
names <- c("archaea", "bacteria", "viral", "fungi", "protozoa","total")
names(names) <- names
#summary files
tables <- lapply(paste0("mini1-", names[1:5], ".out.txt"), read.kraken)
names(tables) <- names[1:5]
total <- as.data.frame(Reduce(function(x,y){ x[,-4] + y[,-4]},
tables, init= matrix(data = 0, nrow = 6, ncol = 2)))
total$rank <- row.names(total)
tables$total <- total
##melt data
mtable <- lapply(tables, melt, id.vars = "rank")
#order levels
levs <- c("ph", "cl", "or", "fm", "ge", "sp")
mtable <- lapply(mtable, function(m) {m$rank<- factor(m$rank, levels = levs); m})
big_table <- do.call(rbind,
lapply(names, function(n) {m <- mtable[[n]]; m$Kingdom=n; m}))
big_table$Kingdom <- factor(big_table$Kingdom, levels = c("archaea", "bacteria",
"viral", "fungi", "protozoa", "total"))
#plots
##plots <- lapply(names, function(n){
## ggplot(data = mtable[[n]], aes(x = rank, y = value, fill = variable)) +
## geom_col() + guides(fill=guide_legend(title="Correct\nClassification?")) +
## labs(title = n, x = "Taxonomic level", y = "Identification rate", label="label")
##})
##ggarrange(plotlist = plots)
#better plot
#pdf(file = "Kraken-summary.pdf", width = 16*2/3, height = 9*2/3)
ggplot(data = big_table, aes(x = rank, y = value, fill = variable)) +
geom_col() + guides(fill=guide_legend(title="Correct\nClassification?")) +
labs(x = "Taxonomic level", y = "Number of Samples", label="label") +
facet_wrap("Kingdom", scales = "free_y")
#dev.off()
##plot2
#make chart from output of krona
FILE_NAME=$1
CUT_O=$FILE_NAME.chart.html
cat $FILE_NAME | cut -f2,3 > $CUT_O | ktImportTaxonomy $CUT_O -o $CUT_O