{kind=link}
![]()
A pipeline for subsampling genomic data based on epidemiological time series data.
If you use this tool in a publication, please cite this DOI: 10.5281/zenodo.7065455
Alternatively, you can cite the original manuscript:
Alpert, T., Brito, A. F., Lasek-Nesselquist, E., Rothman, J., Valesano, A. L., MacKay, M. J., ... & Grubaugh, N. D. (2021). Early introductions and transmission of SARS-CoV-2 variant B.1.1.7 in the United States. Cell, 184(10), 2595-2604.
subsampler runs on MacOS and Linux. To run all steps until the actual subsampling, besides having conda and the subsampler environment installed (see next section), you need to provide the following files:
- Metadata file containing columns with at least: sample names (
strain, oraccession number),date, and geographic columns (country,division, etc). - Matrix of daily case counts per geographic location (matching the geographic level of interested, included in the metadata)
-
If you only need to run this pipeline to calculate the proportion of sequenced cases per geographic location, per unit of time, you just need to run the pipeline up to the
correct_biasstep (snakemake correct_bias). It will produce a matrix with the proportions of sequenced cases. -
For this pipeline to run up to its last step (
snakemake subsample), the metadata file itself must contain the minimum set of columns (described above), correctly named in the Snakefile (here). -
The complete run of
subsamplergenerates, among other files, a TXT file containing a list of accession numbers (gisaid_epi_isl, for example) or genome names (strain, for example), provided the corresponding columns are found in the metadata file used as input. Such list of genome names or (especially) accession numbers can be used to download an actual sequence file from a genomic database (GISAID, for example), so that further analyses can be performed.
git clone https://github.com/andersonbrito/subsampler.git
cd config
conda env create -f subsampler.yaml
conda activate subsampler
To update the conda environment, run:
cd config
conda env update -f subsampler.yaml
Alternatively, mamba can also be used to install the subsampler conda environment.
 Figure 1. Workflow Overview
Figure 1. Workflow Overview
subsampler can perform subsampling using epidemiological data from any geographical level (per country, per states, etc) provided daily case counts are available. See more details in the 'Execution' section. To prepare
- Download and provide a daily case data file
- Generate matrix of case counts, per location (Y axis), per day (X axis)
The pipeline will perform these actions:
- Read genomic metadata file
- Convert date format to YYYY-MM-DD
- Generate matrix of genome counts, per location (Y axis), per day (X axis)
The pipeline will perform these actions:
- Combine genomic and case counts per unit of time (week, month, or year)
- Drop data from time periods outside the boundaries defined by
start_dateandend_date.
The pipeline will perform these actions:
- Read matrices with case and genome counts
- Generate matrix reporting the observed sampling proportions per unit of time
- Generate matrix reporting the sampling bias (under- and oversampling) given the baseline defined by the user
- Generate matrix with the corrected genome count per unit of time, given the pre-defined baseline sampling proportion
The pipeline will perform these actions:
- Read metadata and corrected genomic count matrix
- Read lists of genomes to be kept or remove in all instances (if provided)
- Read the filter file, to include or exclude genomes from certain metadata categories
- Perform subsampling guided by case counts per unit of time
- Generate a list subsampled sequences, and a corresponding metadata file
- Generate a report with number of sampled genomes per location
To run this pipeline, users need to provide a TSV file of daily case counts similar to the format below:
Global case counts
| code | country | 2021-01-01 | 2021-01-02 | 2021-01-03 | 2021-01-04 | 2021-01-05 | ... |
|---|---|---|---|---|---|---|---|
| ABW | Aruba | 20 | 23 | 32 | 42 | 110 | ... |
| AFG | Afghanistan | 0 | 0 | 0 | 1485 | 94 | ... |
| AGO | Angola | 15 | 40 | 34 | 42 | 72 | ... |
| AIA | Anguilla | 0 | 0 | 2 | 0 | 0 | ... |
| ALB | Albania | 0 | 675 | 447 | 185 | 660 | ... |
| AND | Andorra | 68 | 49 | 26 | 57 | 59 | ... |
| ARE | United Arab Emirates | 1856 | 1963 | 1590 | 1501 | 1967 | ... |
| ARG | Argentina | 4080 | 5240 | 5884 | 8222 | 13790 | ... |
| ARM | Armenia | 329 | 60 | 229 | 193 | 324 | ... |
| ... | ... | ... | ... | ... | ... | ... | ... |
Country-level case counts
| code | state | 2021-01-01 | 2021-01-02 | 2021-01-03 | 2021-01-04 | 2021-01-05 | ... |
|---|---|---|---|---|---|---|---|
| AK | Alaska | 5 | 802 | 297 | 264 | 200 | ... |
| AL | Alabama | 4521 | 3711 | 2476 | 2161 | 5498 | ... |
| AR | Arkansas | 4304 | 2000 | 2033 | 1306 | 4107 | ... |
| AS | American Samoa | 0 | 0 | 0 | 0 | 0 | ... |
| AZ | Arizona | 10060 | 8883 | 17234 | 5158 | 5932 | ... |
| CA | California | 39425 | 50222 | 37016 | 38256 | 38962 | ... |
| CO | Colorado | 3064 | 2011 | 2078 | 2185 | 3458 | ... |
| CT | Connecticut | 0 | 4412 | 0 | 4516 | 2332 | ... |
| DC | District of Columbia | 269 | 257 | 255 | 140 | 262 | ... |
| ... | ... | ... | ... | ... | ... | ... | ... |
Using one of the commands below, users can download reformatted daily case count files automatically from CSSE at Johns Hopkins University:
Global data
python scripts/get_daily_matrix_global.py --download yes
US data
python scripts/get_daily_matrix_usa.py --download yes
Users can provide their own daily case count file, as long as it matches the format above (tab-separated, with daily counts, and a column with unique identifiers). If one of the commands above is used, the reformatted matrix of case counts would need to be placed inside /data, and should be named here.
Now, edit the Snakefile to fix the following lines:
-
start_date = "YYYY-MM-DD" (select the start date according to your needs)
-
end_date = "YYYY-MM-DD" (select the end date according to your needs)
-
extra_columns = second column with identifier, such as region, continent (a column found in the original metadata file, which you want to see displayed alongside the
geo_columnin the final outputs)
The subsampler pipeline allows users to calculate the percentage of sequenced cases per location. It aggregates both genome counts and case counts per unit of time, per location (country or state), and proceed with calculations (genomes/cases) to get a time series of proportions of sequenced cases, information useful for monitoring how genomic surveillance is going in different locations.
To that end, the user needs to provide a metadata matrix, similar to the one used by nextstrain, which can be downloaded from GISAID, under Downloads > Genomic Epidemiology. Add the name of that file here, place it inside /data, and run the pipeline up to the correct_bias step using the command below:
snakemake correct_bias
After a few minutes, among the files in /outputs, users will find three matrices, one of them showing the weekly proportion of sequenced cases:
matrix_cases_unit.tsv
matrix_genomes_unit.tsv
weekly_sampling_proportions.tsv
To obtain a list of genomes sampled based on case counts, the last step of the pipeline need to be executed:
snakemake subsample
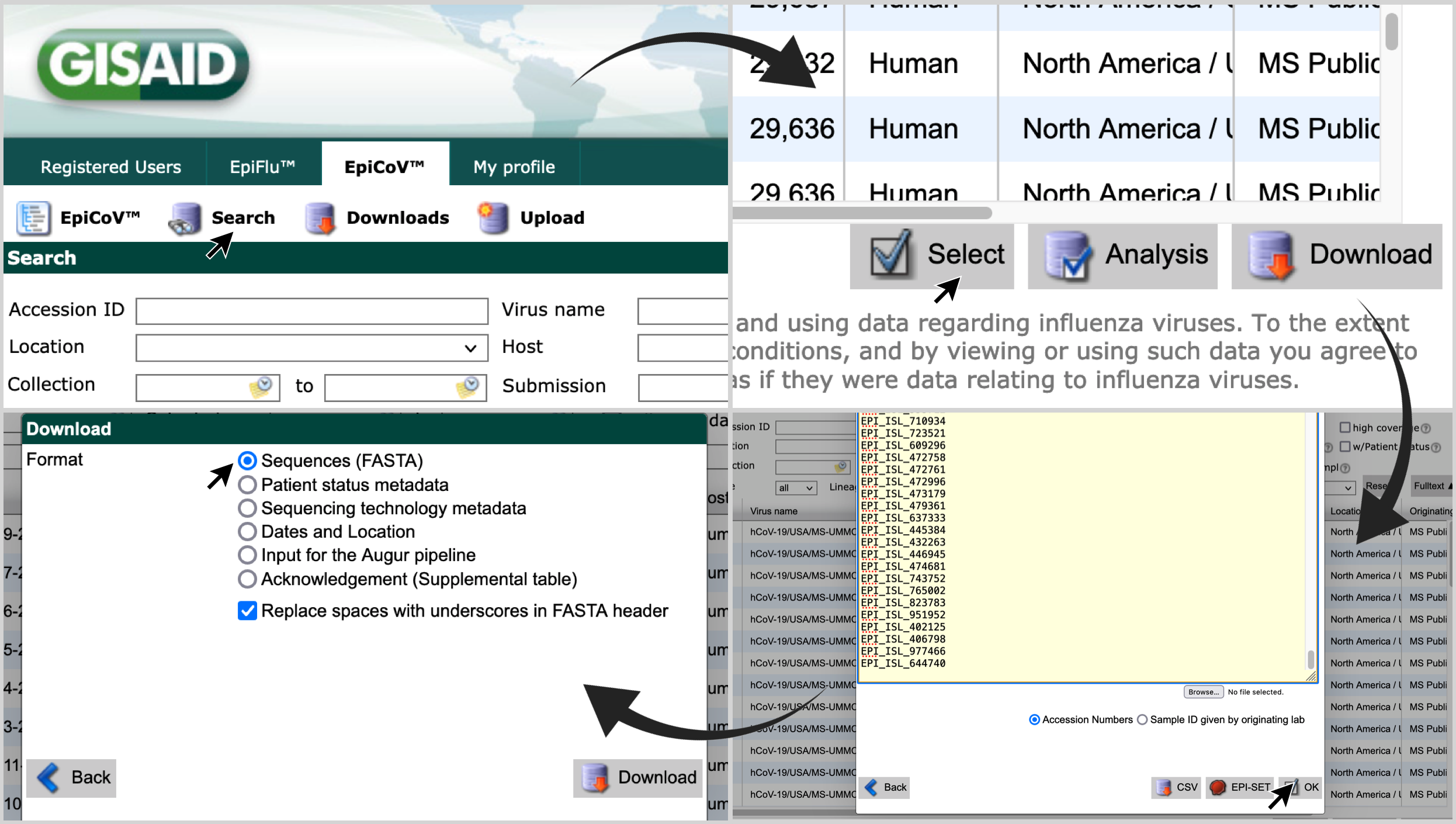
One of the outputs of this pipeline is selected_sequences.txt. If id_column is set here as gisaid_epi_isl, a list of accession numbers of subsampled genomes will be generated. Using that list, proceed as follows:
- Go to gisaid.org; and visit the 'Search' page of 'EpiCov'.
- Click on 'Select'; paste the list of accession numbers in the search box; click on 'OK'; and choose the format 'Sequences (FASTA)'.
Given the observed number of reported cases in each unit of time (week, month, etc), subsampler attempts to normalized the distribution of genomes sampled over time, following a pre-defined proportion of sequenced cases, a baseline defined by the user (here). The scales of the outbreaks worldwide, however, differ by many orders of magnitude, what poses an extra challenge when it comes to subsampling in proportion to reported cases: while a country with 10 million inhabitants may report a few thousand cases per week, larger countries (with populations greater than 100 million) may report more than a million cases in a week. Such large scale may overwhelm the representation of smaller countries in the final dataset, especially if the baseline is not carefully adjusted.
Let's take a look at an example involving the USA (329,5 million inhabitants). Below we have the observed numbers of reported cases and the number of sequenced cases earlier in the pandemic. Under scenarios simulating different proportions of sequenced cases, hundreds or even thousands of genomes may be sampled from each week.
| Country | Data type | 2020_EW10 | 2020_EW11 | 2020_EW12 | 2020_EW13 | 2020_EW14 |
|---|---|---|---|---|---|---|
| United States | Observed number of reported cases | 378 | 2575 | 23047 | 101392 | 192359 |
| United States | Observed number of genomes | 358 | 1695 | 4135 | 3162 | 771 |
| United States | Expected number of genomes under a scenario of 0.1% sequenced cases | 0 | 3 | 24 | 102 | 193 |
| United States | Expected number of genomes under a scenario of 1% sequenced cases | 4 | 26 | 231 | 1014 | 1924 |
| United States | Expected number of genomes under a scenario of 5% sequenced cases | 19 | 129 | 1153 | 5070 | 9618 |
However, under the same parameters shown above, when we look at scenarios in a country with a smaller population, for example, the United Arab Emirates (9.9 million inhabitants), the number of sampled genomes in a given week may be a few dozen, or none at all, as the baseline may be too low to allow sampling of even a single genome. For example, 0.01% sequenced cases in a week with 7,000 reported cases would suggest the sampling of 0.7 genomes, which cannot be performed. In this situation, the expected number of genomes is set to zero. Below we have an example showing the observed numbers of reported cases and the number of sequenced cases in the United Arab Emirates, and the resulting subsampling in different scenarios (see the expected number of genomes in rows where the baseline was set as 0.1 or 1% sequenced cases).
| Country | Data type | 2020_EW10 | 2020_EW11 | 2020_EW12 | 2020_EW13 | 2020_EW14 |
|---|---|---|---|---|---|---|
| United Arab Emirates | Observed number of reported cases | 24 | 40 | 68 | 315 | 1037 |
| United Arab Emirates | Observed number of genomes | 0 | 8 | 5 | 5 | 7 |
| United Arab Emirates | Expected number of genomes, under a scenario of 0.1% sequenced cases | 0 | 0 | 0 | 0 | 2 |
| United Arab Emirates | Expected number of genomes, under a scenario of 1% sequenced cases | 0 | 0 | 0 | 4 | 11 |
| United Arab Emirates | Expected number of genomes, under a scenario of 5% sequenced cases | 2 | 2 | 4 | 16 | 52 |
In summary, given the differences in outbreak scales, depending on the research questions, the user should set up the baseline accordingly, to allow sampling from specific locations or time periods (for example, early phases of an epidemic, where smaller number of cases are reported).
If your questions are not directly related to phylogeography, the subsampler approach (to obtain subsets of genomes sampled based on case counts) may not be what you need. Since the sampling is weighted by case counts, subsampler is more likely to sample genomes from heavily impacted countries (those with more reported cases), and the lower the baseline (the percentage of sequenced cases), the less likely would it be for countries facing small scale outbreaks to be represented (for example, the least populated countries), which end up being overshadowed by larger countries, which may report hundred thousands of cases per week.
If you are not trying to infer ancestral states in a phylogeographic perspective, but is more interested in phylodynamic questions, subsampling based on the timing of the events (waves, introductions, seasons, etc) is a better approach.
In this repository you can find genome_selector.py, a python script designed to sample genomes without taking into account case counts, but instead, following specific variables (date of colection, country, viral lineage, or any other metadata column). For example, the table below illustrates a sampling scheme to obtain around 650 genomes of viruses belonging to lineage B.1.1.7 (Alpha variant), circulating in the US and the UK, between 2020-12-01 and 2021-06-30, with other US and European samples as contextual genomes. In this hypothetical example, note that US contextual genomes are selected from two time periods, and in different proportions: 50 genomes up to late November 2020, and 100 genomes from December 2020 onwards (from any lineage). Following these criteria, genomes will be sampled per week, and the number of genomes in each week will be proportionally defined by the number of available genomes in each week, according to the parameters defined in each row of the sampling scheme file.
Also, the scheme is set up to ignore genomes from California and Scotland: genomes from those locations will not be included in any instance, since they are filtered out prior to the genome selection step. To reproduce the scheme below, genome_selector.py will use a --metadata file listing all genomes from the locations and lineages represented below.
genome_selector.py is not part of subsampler. It should be executed separately:
python genome_selector.py --metadata METADATA --scheme SCHEME
... where --scheme is a TSV file with this format:
| purpose | filter | value | filter2 | value2 | sample_size | start | end |
|---|---|---|---|---|---|---|---|
| focus | pango_lineage | B.1.1.7 | country | USA | 200 | 2020-12-01 | 2021-06-30 |
| focus | pango_lineage | B.1.1.7 | country | United Kingdom | 200 | 2020-12-01 | 2021-06-30 |
| context | region | Europe | 100 | 2020-12-01 | 2021-06-30 | ||
| context | country | USA | 50 | 2020-11-30 | |||
| context | country | USA | 100 | 2020-12-01 | |||
| ignore | division | California | |||||
| ignore | division | Scotland |
Among the outputs of genome_selector.py, users will find text files containing the list of around 650 genomes, both as names (e.g. USA/CT-CDC-LC0062417/2021) and as accession numbers (e.g. EPI_ISL_2399048). The last file can be used to filter and download genomes directly from gisaid.org, as explained above.
v1.1.0 / 2022-06-12:
- Fasta file with the actual sequences are no longer required as input file. Now, by default, the pipeline will not inspect the level of completeness of the genomes, but will focus on subsampling based on metadata rows only. However, asessement of sequence quality is still supported.
- A
filter_fileis now an input of this pipeline (see 'config/filters.tsv', and this line. With this file, users can determine specific data categories to be included or excluded. This feature is useful, for example, for subsampling 'variant-specific' data (e.g. include → pango_lineage → B.1.1.7), among other uses. - Users can now specify the time unit of the time series (week, month or year), which should be set according to the most adequate time period that match the evolutionary time scale of the viruses under study (for SARS-CoV-2, 'week' is an adequate option, but for Dengue virus, 'month' is the best option).