Variable Length Hidden Markov Models (VLHMM)
The repository concludes two modules vlhmm, chipseq, which can solve next problems:
- vlhmm:
- building, training, plotting context transition tree
- building, training VLHMM
- chipseq:
- applying VLHMM to ChIp-seq data
- conversion for genome browser fromat http://genome.ucsc.edu/
Language: Python 3.x
Dependencies:
- Cython
- NumPy, SciPy
- Pylab
- datrie
- PyGraphviz
Instalation:
git clone https://github.com/atanna/hmm.git
Cython-files compilation:
python setup.py build_ext --inplace
The directory vlhmm_/examples/ concludes test examples with training VLHMM and context trees on simulated data.
chipseq/real_test.py shows example VLHMM on ChIP-seq data
- Context transition tree defines a stochastic process.
- Context state -- any preffix from previous states (the process moves from right to left, i.e. states go in descending order of time).
- Vertex ~ context.
- Edge ~ state.
- Outdegree of internal vertex -- number of states.
- The leaf defines the distribution of the current state.
-
-
"Unfair Coin"
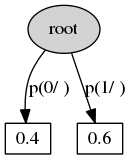
-
Markov chain
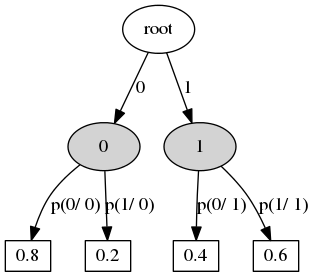
-
Second-order Markov chain
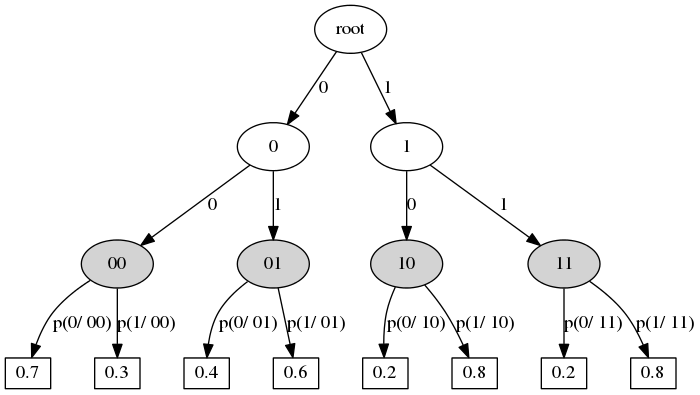
-
Variable length Markov chain
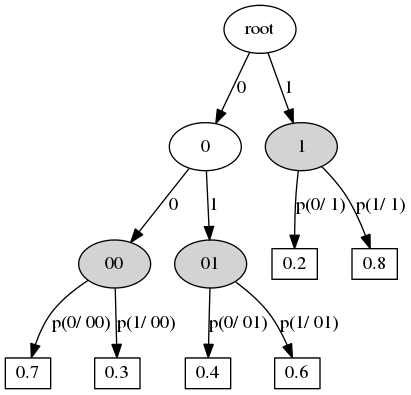
-
*Context transition tree defines a variable leghth Markov stochastic process.
- The same as HMM.
- Hidden layer is defined by variable leghth Markov stochastic process (which we can define by context transition tree).