Benchmark Dose Analysis
Benchmark dose analysis consists of fitting dose-response data to a collection of parameterized equations (models), followed by choosing the model that best describes the data while minimizing complexity (i.e., the best fit model). Alternatively, non-parametric (GCurveP) modelling may be performed.
To perform BMD computations, select complete or filtered data set(s) from the Data Selection Area, and click Tools > Benchmark Dose Analysis. Then choose either EPA BMDS Models (Parametric), or Sciome GCurveP (Non-parametric).
Video describing Benchmark Dose Analysis setup
Document describing model inputs and outputs, and of the best model selection work flow
Document describing GCurveP method and work flow
BMDExpress 2 is available for Windows, Mac and Linux operating systems, however BMD results may be slightly different across these operating systems due the floating point calculations employed by the BMDS model executables that are used in BMDExpress. The model executables were originally intended to run on Windows operating systems and are therefore only validated in Windows. Empirical evaluation of the differences across platforms indicate there are minimal changes to the results, however the results from a limited number of probes may change significantly.
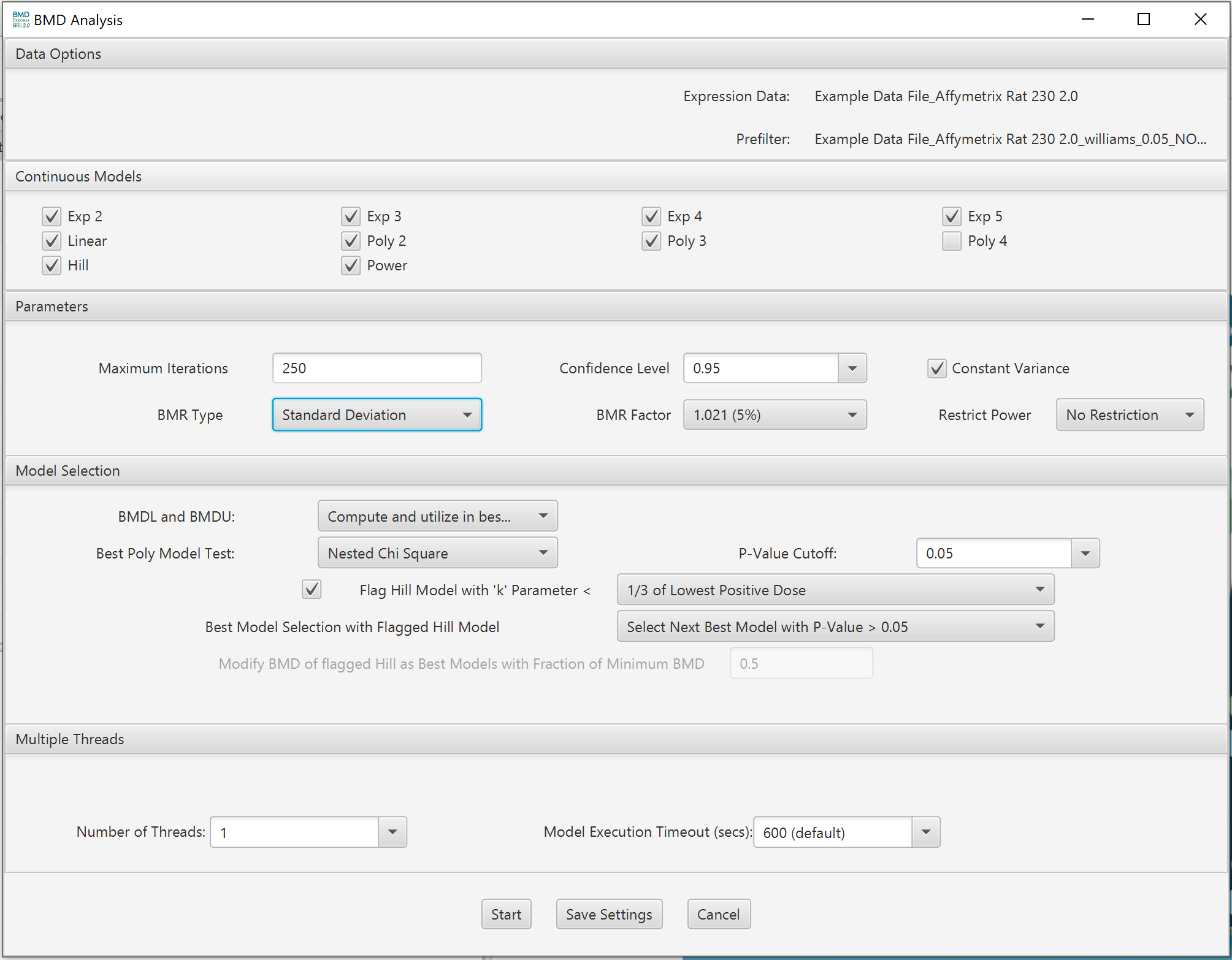
Lists expression data and any pre-filtering in the current workflow.
Choose model(s) for curve fitting. Some of the models will be selected by default. The number of polynomial models available is automatically determined by the unique number of doses comprising the dose-response data. Any single model or combination of models may be chosen.
Model Equations for continuous models implemented in BMDExpress 2.0 are shown below, and described in greater depth in the BMDS User Manual. For all equations, µ is the mean response predicted by the model.
-
Polynomial model:
μ(dose)=β_0+β_(1 ) dose+ β_(2 ) dose^2+ ⋯ + β_(n ) dose^nwhere n is the degree of the polynomial. -
Linear model: The linear model is a special case of the polynomial model with n fixed at 1.
-
Power model:
μ(dose)=γ+ β dose^δwhere 0 < γ < 1, β ≥ 0, and 18 ≥ δ > 0. -
Hill model: μ(dose)=γ+ (v dose^n)/(k^n+ dose^n )
-
Exponential 2: μ(dose)=aexp(sign b*dose)
-
Exponential 3: μ(dose)=aexp(sign (b*dose)^d)
-
Exponential 4: μ(dose)=a*(c-(c-1)exp(-1 b*dose))
-
Exponential 5: μ(dose)=a*(c-(c-1)exp(-1 (b*dose)^d ))
Note: For the the exponential 2 and 3 models, "sign" is the adverse direction
Note2: Exponential models are intolerant of negative data values, hence if the user plans to use the models data must be normalized accordingly.
Note3: Exponential and Power models require atleast 2 biological replicates at each dose level in order to be fit to the data. Other models (Hill and polynomial models) will run when there is only one replicate at each dose level.
Curve fitting is performed using methods implemented in the U.S. Environmental Protection Agency’s BMDS. Adverse direction is determined automatically in the curve fitting computation (i.e., determined by the linear trend test embedded in the model). Parameters that cannot be altered by the user are: benchmark response (BMR) type (set to Std Dev), relative function convergence (set to 1e-8), parameter convergence (set to 1e-8), BMDL curve calculation (set to 1), BMDU curve calculation (set to 1) and smooth option (set to 0).
- Maximum Iterations: A convergence criterion for the model.
- Confidence Level: The statistical lower confidence limit applied to the BMD estimated by the model. The resultant lower bound on the BMD is the benchmark dose lower confidence limit (BMDL).
- Constant Variance: When selected an assumption of constant variance is used in the modeling.
- BMR Type: Standard deviation or relative deviation.
- BMR Factor: Also called the benchmark response or critical effect size in some publications, the number of standard deviations at which the BMD is defined. The BMR is defined relative to the response at control. Since both the response at control and the standard deviation used to calculate the BMR are parameters estimated as part of the curve fit, the BMR may change when the model used to fit the data changes. The recommended default BMR factor is 1 (equivalent to 1 standard deviation), consistent with EPA recommendations for continuous data.
-
Restrict Power: The parameter “Restrict Power” is only applied to Power model. It allows the user to restrict the power parameter to be >= 1. The default setting is restricting the power to >= 1.
- Note: For all exponential models and the Hill model power is restricted to be >= 1. For the linear and poly models power is a fixed value defined by the degree of the polynomial.
-
BMDL and BMDU: Statistical lower and upper bounds on the computed benchmark dose. Choose whether to compute, and whether to include in best model selection criteria.
-
Best Poly Model Test:
- Nested Chi Square: A nested likelihood ratio test is used to select among the linear and polynomial (2° polynomial, 3° polynomial, etc.) models followed by an Akaike information criterion (AIC) comparison (i.e., the model with the lowest AIC is selected) among the best nested model, the Hill model and the power model.
- Lowest AIC: A completely AIC-based selection process is performed.
-
P-Value Cutoff: Statistical threshold for the Nested Chi Square test when selecting the best linear/poly model
-
Flag Hill Model with ‘k’ Parameter < : If the Hill model is selected as one of the models to fit the data, flag a Hill model if its ‘k’ parameter is smaller than the lowest positive dose, or a fraction (1/2, or 1/3) of the lowest positive dose. This option is included since the Hill model can provide unrealistic BMD and BMDL values for certain dose response curves even when it provides the lowest AIC value. For flagged Hill models, there are multiple options when selecting the best model:
- Include Flagged Hill Models: There is no additional condition applied when selecting the best model.
- Exclude Flagged Hill from Best Models: The flagged Hill model will not be considered when selecting the best model.
- Exclude All Hill from Best Models: All Hill models, either flagged or not, will not be considered when selecting the best model.
- Modify BMD if Flagged Hill as Best Model: If a flagged Hill model is selected as the best model, then modify its BMD value based on a defined fraction of the lowest BMD from the feature/probe set in the data set with a non-flagged Hill model as the best fit model. Note: With this option the user will need to enter a value in the "Modify BMD of flagged Hill as Best Models with Fraction of Minimum BMD". The default value for this parameter is 0.5. Warning: we do not recommend doing this if the object is to identify individual feature or gene BMD values because this can provide an inaccurate estimate of potency.
- Select Next Best Model with P-Value > 0.05: The next best model will be selected that meets both the minimum AIC value and a goodness-of-fit p-Value > 0.05. If another model can not be identified to replace the flagged Hill model a BMD, BMDL and BMDU value is assigned to the probe that equals 0.5*the lowest BMD, BMDL and BMDU of the best models after excluding the flagged Hill models.
-
Warning: Before selecting the settings in this section the user will want to perform testing to determine the optimal parameter setting to ensure complete model execution. To start the optimization process it is suggested that the user set their threads to no more than 4 times the number of cores available and set the model execution time out to 600 seconds. In windows the number of cores can be found in the "Task Manager" under the "Performance" tab. Once the Benchmark Dose modeling is finished the user will want to check th results table to determine if all models for all features executed to completion (i.e., a "true" value is shown in the "<model name> Execution" column). For details see the tutorial video on Threads and Model Execution Timeout.
-
Number of threads: This option allows the user to perform multiple model fit computations in parallel by utilizing multiple CPU cores. The option increases the efficiency of CPU usage and significantly reduces the computational time. For example, a computer with a quad-core processor will theoretically require 1/4th the time to complete model fitting when 4 threads are selected rather than 1 thread. In practice, the actual efficiency varies. Try different values to optimize in your particular situation. Open a processor monitoring utility, and observe processor utilization. Typically, utilization of >80% can be achieved when setting the number of threads to be several to 10-fold greater than the number of processor cores. The default recommendation for the number of threads is 4 times the number of cores that are available, assuming that the user is employing the recommended 600 second model time out (see Model Execution Timeout). The recommendation is less than this if the user plans to carry out additional computational tasks while modeling is being performed.
- N.B. increasing processor utilization by BMDExpress will hinder interactive responsiveness.
-
Model Execution Timeout (secs): This option determines how long a model is run on an individual probe. Default setting is 600 seconds. The default should be adequate to fit all models without the model timing out before its finished. Notations are added to the results table in Benchmark Dose Analyses under the column heading "<Model Name> Execution Complete". A "True" value indicates the model execution was complete. Note: The user has the option of typing in their own value (i.e., using a value not in the drop down) for model time out.
After selecting and checking the appropriate data, models, parameters, and other options, click Start. Computation may take minutes to hours depending on the total number of probe set identifiers and data sets submitted for analysis, the number of models to fit, and your computer’s performance characteristics.
Statistical outliers in the dose-response data can result in a non-monatonic curve fit. Some users interpret this as an unrealistic outcome. GcurveP is a non-parametric curve correction algorithm that is based on the assumption that the correct dose-response curve must exhibit monotonic behavior. It finds a minimal set of corrections needed to restore monotonic behavior of the dose-response curve. It takes the direction of monotonicity (i.e., ascending, flat, or descending) as its input parameter, and iteratively searches from each dose-group of the curve, as its trusted pivot point, for monotonicity violations. It then counts how many other curve points would need adjustments (i.e., by shifting respective dose groups) to restore monotonicity. The final solution is then the set with minimal number of such points that require corrections.

Lists expression data and any pre-filtering in the current workflow.
-
BMR Factor: Also called the benchmark response or critical effect size. It is a multiple of standard deviation (SD) of the probe/gene expression change relative to its expression level at control. It determines the change in expression at which the BMD is defined. We recommend setting a BMR Factor to 3 when using GCurvep because it uses an adjusted standard deviation that down weights outlier values that leads to a compression of the estimated SD.
-
Bootstraps: Monotonicity correction is repeated by a bootstrap sampling algorithm. 1000 times is the default.
-
pValue Confidence Interval: Default is 0.05, which means the software will calculate a 95% confidence interval
- Number of Threads: Not yet implemented.
Video describing Benchmark Dose Analysis results

Results are tabulated in the bottom half of the window:
- Probe ID: Unique identifier for probe/probe set ID.
- Genes: All genes included in probe/probe set.
- Gene Symbols: Gene symbols included in probe/probe set.
When BMDS is used for curve fitting, there are columns for the "Best" model, and the other models that were computed:
- BMD: Benchmark dose
- BMDL: Lower bound of the 95% confidence interval of the benchmark dose
- BMDU: Upper bound of the 95% confidence interval of the benchmark dose
- fitPValue: Global goodness of fit measure. Small p-values indicate that the model is a poor fit to the data. More specifically, the model identified by AIC is further compared with the dose response from fully saturated model (unconstrained model that is free from any functional form or trajectory constraints) using conventional likelihood ratio test statistic. The larger p-value for this test indicates that the identified model is not statistically inferior fit than the saturated model, whereas the smaller p-value indicates that the identified model does not fit the data as well as the saturated model and hence it is possible to device a better fitting model.
- fitLogLikelihood: A value calculated using the log of the likelihood given the model, used in calculating the AIC. The value is used to compare between different model fits to the same feature.
- AIC: Akaike Information Criterion. Given a set of dose-response models for a probe set/gene the AIC estimates the quality of each model relative to the other models. In most cases with BMDExpress the model with the lowest AIC is selected as the "best model".
- adverseDirection: Direction of the dose-response (i.e., up- or down-regulation) as identified by the software
- BMD/BMDL Ratio
- BMDU/BMDL Ratio
- BMDU/BMD Ratio
- Prefilter P-Value: statistical cutoff in probe selection step
- Prefilter Adjusted P-Value: statistical cutoff in probe selection step, with adjustment applied
- Max Fold Change: maximum fold change of all probes selected for BMD computation
- Max Fold Change Absolute Value
- NOTEL: No Observed Transcriptional Effect Level; highest dose that does not cause a transcriptional change in a transcript/gene.
- LOTEL: Lowest Observable Transcriptional Effect Level; lowest dose resulting in transcriptional change in a transcript/gene.
- FC Dose Level <n>: fold change at each dose level n
- Flagged: Value = "1" indicates that the Hill model was flagged for that probe or gene based on the setting selected in the BMD analysis set up. "0" indicates the Hill model was not flagged
- For each model fit to the data, specific parameters for each probe set/gene are reported. This is done to allow the user to recapitulate the model equations.
- There is also a corresponding set of columns for the "Best" fit model (i.e., BMD, BMDL, BMDU, fitPValue, fitLogLikeihood, AIC, BMD/BMDL, BMDU/BMDL, Prefilter P-value, Prefilter Adjusted P-value, Max Fold Change, Max Fold Change Absolute value),
- FC Dose Level n: Fold change at dose n.
- For a more detailed definition of terms please refer to the EPA BMDS User Guide
In the case of GCurveP, there are columns similar to the BMDS models, plus GCurveP-specific results:
- fitValue: fraction of original signal preserved after GCurveP treatment
- Excecution Complete: true or false; indicates whether computation completed under the selected initial conditions
Each probeset ID is a hyperlink to a separate window that displays a plot of the corresponding dose response behavior and model fit curves.
 The curve that is shown initially is the one with the best fit as described in the introduction, but you can view the fit of other models by using the Model dropdown menu.
The curve that is shown initially is the one with the best fit as described in the introduction, but you can view the fit of other models by using the Model dropdown menu.

The Mean & Standard Deviation checkbox toggles the points in the plot to reflect the mean and standard deviation of each dose, or the response data points.

You can also change the scale of the axis to linear by unchecking the Logarithmic Dose Axis checkbox.

You can switch between different probes/probe sets inside of the individual curve viewer using the ID dropdown, however it is usually faster to close the popup and double-click on a different probe/probe set in the results table.

All properties of the curve can be altered in the Properties Menu, accessed by right-clicking in the chart area.

Inside the Chart Editor for the plot, there are a variety of parameters that can be changed to alter the appearance of the plot.
-
Title
- Show Title
- Title Text
- Font
- Color
-
Plot
- Domain & Range Axis
- Label
- Font
- Paint
- Other
- Ticks
- Show tick labels
- Tick label font
- Show tick marks
- Range
- Auto-adjust range
- Minimum range value
- Maximum range value -TickUnit
- Auto-selection of TickUnit
- Tickunit Value
- Ticks
- Appearance
- Outline Stroke
- Outline Paint
- Background Paint
- Orientation
- Domain & Range Axis
-
Other
- Draw anti-aliased
- Background paint
- Series Paint
- Series Stroke
- Series Outline Paint
- Series Outline Stroke
Finally, you export an image of the plot by right clicking on the image and selecting "Export As" and then selecting the image file format.
The default visualizations are:
-
BMDS Model Counts

-
Best BMD Vs. Best BMDL

-
Best BMD Histogram

Additional visualizations are available by making a selection in the Select Graph View dropdown list.
These parameters are changed via the filter panel. You must also make sure that the Apply Filter box is checked in the toggles panel for these filters to be applied. The filters will be applied as soon as they are entered; there is no need to click any apply button other than the checkbox.
There is a filter available for every column in the BMD results table. Some particularly useful ones are:
- Best BMD: Filter by best BMD
- Best BMD/BMDL: Filter by BMD/BMDL ratio
- Best BMDL: Filter by best BMDL
- Best BMDU: Filter by best BMDU
- Best BMDU/BMD: Filter by BMDU/BMD ratio
- Best BMDU/BMDL: Filter by BMDU/BMDL ratio
- Best Fit Log-Likelihood: Filter by Log-Likelihood
- Best Fit P-Value: Filter by fit p-value
- Gene ID: Filter by gene IDs.
- Gene Symbols: Filter by gene symbols.
