This simple orientation-dependent coarse-grained knowledge-based scoring function for protein-ligand docking developed in a joint-venture with Sergei Grudinin lab outperforms state-of-the-art statistical potentials using a 3D joint probability and a minimalist protein representation. Our side-chain independent potential is a fast and highly discriminative energy function for protein-ligand docking, please have a try. This mirrors the methodology site from here
M. Kadukova, K. dos Santos Machado, P. Chacón, S. Grudinin (2021) KORP-PL: a coarse-grained knowledge-based scoring function for protein-ligand interactions, Bioinformatics, 37(7), 943–950 

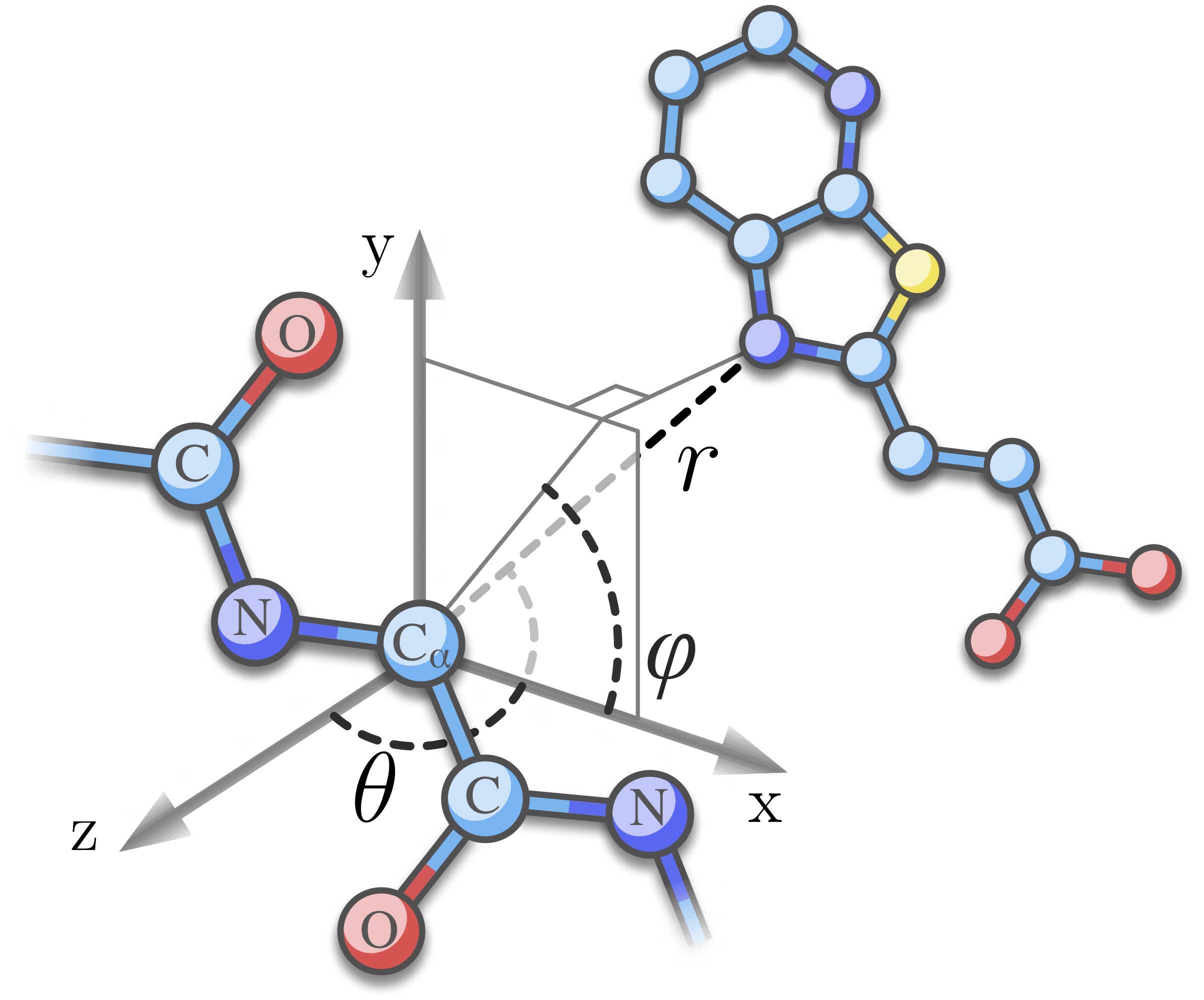
| KORP-PL is a novel sidechain-free coarse-grained knowledge-based scoring function for protein-ligand interactions developed together with Sergei Grudinin. To this end, we first collect sidechain-free statistics of the distances and angles between an oriented frame built on a residue backbone and a ligand atom for each residue and ligand atom within a cutoff distance as depicted in the left figure. Similar framework was successfully used for protein and loop modeling in the 6D orientation-dependent knowledge-based potential KORP[1]. The interaction potentials are then derived in a classical way[2] and keep the full dependence between variables. KORP-PL was trained on PDBBind 2016[3] with ligand atom typization similar to the one from Convex-PL[4]. Our side-chain independent potential is a fast and highly discriminative energy function for protein ligand docking, please try it. |
|
|
Schematic view of the relative orientation of a ligand molecule to a protein residue. The residue is represented with a 3D oriented frame built from three backbone atoms. The relative orientation of a ligand atom is described by two spherical angles, θ, the polar angle between the r and z vectors, and φ, the azimuthal angle between x and the projection of r into the xy plane. |
The usage of KORP-PL is very simple:
>~/korp-pl/korp-pl-test$ ../KORP-PL --receptor ./10gs/10gs_protein.pdb --ligand ./10gs/10gs_ligand.mol2 --mol2 #************************************************************ #*----------------------------------------------------------* #*-------------------------KORP-PL--------------------------* #*-------------------------Authors:-------------------------* #*--------Maria Kadukova, Karina dos Santos Machado,--------* #*------------Pablo Chacon, and Sergei Grudinin.------------* #*-----Copyright (c): Nano-D team, Inria/CNRS Grenoble,-----* #*-------Rocasolano Institute of Physical Chemistry --------* #*-----------France, Russia, Spain, 2019 - 2020.------------* #*-------------e-mail: sergei.grudinin@inria.fr-------------* #*----------------------------------------------------------* #************************************************************ Number of decoys: 1 model 0, energy = -343.3552884724235810
Linux: KORP-PL v0.1 binary and test files macOS KORP-PL v0.1 binary and test files
CASF Benchmarks contain several exercises measuring the pose prediction (docking test), relative affinity prediction (scoring and ranking tests) powers of a scoring function, and a virtual screening exercise (screening test).
CASF Benchmark 2013[5]
We obtained top-ranked results in docking and screening tests. However, our performance in affinity prediction exercises was below the average.
files (CASF Benchmark 2013 rescorings)
CASF Benchmark 2016[6]
We obtained top-ranked results in docking and screening tests. In the screening test evaluated by the enrichment factor metric we outperformed other scoring functions more than 1.5 times. However, our performance in affinity prediction exercises of this benchmark is rather poor.
files (CASF Benchmark 2016 rescorings)
D3R-based benchmark
We compiled a benchmark from the D3R Grand Challenges 2[7], 3[8], and 4[9] user submissions and native structures. It contains pose prediction and scoring exercises. The pose prediction test aims to find a near-native pose among user submissions from each of the Challenges (i.e. it does not require manual docking). Scoring test measures the correlation between predicted binding affinities and experimental constants of co-crystal complexes from the Challenges.
KORP-PL showed very good results in all the three D3R pose prediction tests. Among them, it perfectly performed in the test derived from the Grand Challenge 3, which was a tough case for all the non-template-based docking protocols during the challenge timeframe owing to the wide binding pocket hydrophobicity and high chemical diversity of ligands. As for affinity predictions, KORP-PL obtained very nice results for the Grand Challenge 2 and 4 targets, but a near-zero correlation for the Grand Challenge 3.
files (all benchmark files + rescorings)
[1] Lopez-Blanco, J. R. and Chacon, P.. KORP: knowledge-based 6D potential for fast protein and loop modeling. Bioinformatics, 2019
[2] Samudrala, R. and Moult, J.. An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J. Mol. Biol., 1998
[3] Wang, R., et al. The PDBbind database: methodologies and updates. J. Med. Chem. (2015) link
[4] Kadukova, M. and Grudinin, S.. Convex-PL: a novel knowledge-based potential for protein-ligand interactions deduced from structural databases using convex optimization. J. Comput. Aid. Mol. Des., 2017
[5] Li, Y., et al. Assessing protein–ligand interaction scoring functions with the CASF-2013 benchmark. Nature protocols, 2018, link
[6] Su, M., et al. Comparative assessment of scoring functions: the CASF-2016 update. J. Chem. Inf. Model., 2019, link
[7] Gaieb, Z., et al. D3R Grand Challenge 2: blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput. Aid. Mol. Des., 2018, link
[8] Gaieb, Z., et al.. D3R Grand Challenge 3: blind prediction of protein–ligand poses and affinity rankings. J. Comput. Aid. Mol. Des., 2019, link
[9] Parks, C. D., et. al. D3R Grand Challenge 4: blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput. Aid. Mol. Des., 2020, link