Proposed API for DualTopologyFactory #21
Comments
|
This is the next thing on my list for https://github.com/choderalab/perses, but I think it can wait until after the R01. Any input from @pgrinaway or @andrrizzi? |
|
Looks great to me. As for the questions, I like the idea of specifying functions (maybe with default |
|
Here was the original question:
What if we have an option, like slave_functions = {
'lambda_sterics' : 'lambda',
'lambda_electrostatics' : 'lambda',
...
}By default, then, you only have to control a global Do we like the original factory API? or maybe we don't even need a factory object if it just outputs a single alchemically perturbed system? |
|
Tagging @pgrinaway for additional input. |
Instead of handling this with two different arguments ( |
|
I think the slave_functions = {
'lambda_sterics' : 'lambda',
}If you want to control things with two different functions, 'lambda_sterics' : 'lambda1',
'lambda_electrostatics' : 'lambda1',
'lambda_bonds' : 'lambda2',
'lambda_angles' : 'lambda2',
'lambda_torsions' : 'lambda2',
}If you want to slave everything to 'lambda_sterics' : 'lambda',
'lambda_electrostatics' : 'lambda',
'lambda_bonds' : 'lambda',
'lambda_angles' : 'lambda',
'lambda_torsions' : 'lambda',
}If you want to control everything independently, use a blank |
|
Oh, I think I see what you mean. Do you have in mind something like slave_functions = {
'lambda_sterics' : 'lambda1',
'lambda_electrostatics' : 'lambda1',
'lambda_bonds' : 'lambda2'
}
kwargs = {
'lambda1': [1.0, 0.9, ..., 0.0],
'lambda2': [1.0, 0.5, 0.0]
}
DualTopologyFactory(..., slave_functions, **kwargs)? If this is what you mean, I like it a lot. What I would like to avoid is a signature like DualTopologyFactory(..., slave_functions, lambda_sterics=None, lambda_electrostatics=None, ...)where multiple arguments ( |
|
Ah, I understand now! We definitely want to avoid that.
This function just sets up a System with some context parameters that can
be changed on the fly. By default, each force class ends up with a separate
lambda parameter. By providing these functions, we can insert additional
terms into the energy functions that slave some or all of these parameters
to some user specified global context parameters, which can be useful if
you want to control multiple forces at once. Otherwise you would need to
specify all the lambdas every time.
No values are ever passed. They are always just set to the default of 1.0.
So no additional value arguments are needed.
|
|
Ah! I completely misunderstood how the Then, I like the
If we want to drop factories and adopt that design, we need to transform |
|
Tagging @ppxasjsm. |
|
A variety of questions/comments: First, are you guys discussing somewhere (here or elsewhere) why dual topology as opposed to single topology? Unless you're using the terminology differently than I've seen it in the literature, dual topology means effectively having two separate ligands/solutes at the same time, and you would (if following typical literature approaches) apply some sort of restraints to keep these on top of one another. I've had some problems with restraints when exploring these types of calculations, and have mostly been doing single topology relative free energy calculations. This is also what Schrodinger and others are doing these days. Dual topology also makes some things (such as residue mutations in proteins) a lot more tricky to set up. If this is an unrelated issue, please point me to the appropriate thread. Related to what's here: @jchodera wrote:
I would argue for full control. Optimal ways to do these is still very much a research problem so we want to allow as much control as possible. However:
This seems to be a reasonable solution which, as I read it, allows both worlds. Many users will likely want to control these according to some function, so this allows that AND allows people to set them independently if desired. If indeed dual topology, you also may have the issue that you want to allow what I'd call a "reversible" lambda schedule - i.e. whatever you're doing to system 1, you want to do in reverse to system 2. So for example if you have a lambda_electrostatics which goes from (0, 0.25, 0.5, 0.75, 1.0, ..... 1.0) while lambda_sterics goes from (0, 0, 0, 0, 0, 0.25, ..... 1.0) (that is, you first change charges and then change sterics -- currently we're seeing that even with soft-core Coulomb this may be needed in some cases) you would want this to apply to the transformation of the solute/ligand in system 1 into dummy atoms, but you would want the reverse protocol (first change sterics and then change charges) to apply to the transformation of the solute/ligand in system 2 into dummy atoms.
As mentioned in the OpenFF discussion, there might be a scenario for using some amount of soft core to improve sampling of liquids, for example. While that's not directly relevant to relative calculations as discussed here, I'd argue that things like that are reasons to keep these type of parameters as alchemical parameters that the user can change. Reasonable defaults should be set at system creation time, I believe. Otherwise, this looks reasonable to me. |
I think there is some confusion about the terminology. It's really a hybrid form:
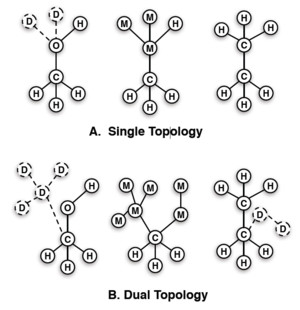
I believe this is what you mean when you are talking about single-topology. You are probably not doing a true single-topology method because you would be changing the topology of the chemical graph and potentially drastically altering the hybridization state of individual atoms. (Did you make this figure?) |
|
We can definitely have softcore Coulomb capability here, with the option of turning it off to get regular Coulomb interactions simply by setting |
Ah, OK. So I call that "single topology", though maybe "hybrid" is OK too. But it's not "dual topology" - in "dual topology" (which is what NAMD has), as I understand it, you can't actually do things like change atom types and so on. As I understand it, if you want to do something like replace ARG with LYS for a sidechain in a protein in NAMD (or with dual topology), you have to actually set things up to turn off the ARG sidechain while turning on the LYS sidechain, and if you want them to occupy the same space you need to restrain them to sit on top of one another. The name "dual topology" as I understand it means that you are tracking the positions of all of the atoms of both entities at the same time. Maybe the distinction is blurry though, as now that I think about it more, in this case the entities are residues and so we're really only dealing with PARTS of a molecule... We actually did some testing at one point where we were trying to use this for absolute hydration free energy calculations and it ended up becoming a horrible mess. Specifically, the restraints people apply to keep the objects in the same space seemed to end up distorting them in a way that led to incorrect free energies at least in some cases.
That's not my figure. And in fact both of those are what I call "single topology" calculations - they just differ in that in (A) one of the end states has no dummy atoms. Again, this is probably all just terminology. The definition I've been using for single topology vs dual topology is shown in these images.
We've found the AMBER version is terrible, as well. Also we haven't had much more success with the GROMACS version. Really we've only had any success (at least for "significant" transformations which involve turning a reasonable sized group into dummy atoms while also turning another reasonable-sized group from dummy atoms into interacting) with regular Coulomb. Have you had any success with softcore? |


{kind=link}
I don't believe there is a way to do this correctly for polymeric molecules unless you also break/form bonds in the process. For small molecules, you could do this, which is what our "multitopology" code does: replicate every atom---even shared ones---and restrain or constrain them to be on top of each other.
Good to know. In principle, the free energies should be correct, but I imagine there could be issues with the variance being large unless constraints are used. OpenMM provides a way to use virtual sites for this purpose. We haven't had much success with softcore Coulomb for absolute free energy calculations yet, but I believe this is due to deficiencies in the parameterization of the functional form and a failure to link the lengthscales to the same lengthscales involved in the softcore Lennard-Jones. I think there are some good ideas now (including those from @mrshirts) on how to automatically optimize these paths, so I'm looking forward to playing with this more. |
|
Steinbrecher had a paper on the constraints required to get SC coulomb and http://onlinelibrary.wiley.com/doi/10.1002/jcc.21909/full On Tue, Apr 19, 2016 at 7:27 PM, John Chodera notifications@github.com
|
We need the ability to create OpenMM
Systemobjects with context parameters that allow us to perform relative free energy calculations between twoSystemendpoints.We presume that the user starts with:
Systemobject for system 1Systemobject for system 2Proposed API
How does this look for an API:
Here
atom_mappingis a dict that maps atoms insystem1tosystem2.We can add some other optional arguments to the factory:
context_parameter_prefixthat allows us to specify the prefix prepended to alchemical context parameters (with the default oflambda, e.g.lambda_sterics,lambda_electrostatics); this could avoid parameter namespace collisions if other alchemical components are in use (e.g. alchemical softening of a loop or residues near a binding site through an orthogonal parameter)alpha(Lennard-Jones) andbeta(Coulomb) softcore parametersSome questions:
lambda_sterics,lambda_electrostatics,lambda_torsion,lambda_bonds,lambda_angles), or do we want to have a singlelambdaparameter and let the user specify a set of functions to describe how these various context parameters are slaved to the single controlling function?alpha,beta) and various exponents, could either be hard-coded atSystemcreation time, could be left as alchemical parameters the user could change, or could have functions specified that describe how they are slaved to a masterlambdaparameter. Which one would be more useful?Implementation details
The current concept is to follow closely the create_relative_transformation code from
examol.All
Forces must appear in the same order in each system. Failure to do so will trigger an exception.Each
Forceterm in is handled differently:HarmonicBondForceandHarmonicAngleForceBonds and angles present in both
system1and system2are converted toCustom*Forces. Force constants (K) and equilibrium values (r0,theta0) are linearly interpolated between their initial and final values according tolambda_bondsandlambda_angles`.PeriodicTorsionForceBecause the number and periodicity of Fourier terms can differ between
system1andsystem2, it seems most appropriate to havelambda_torsionsproduce a linear interpolation of the energies of the two systems.NonbondedForcealphaandbetacontrolling this.GBSAOBCForceThis will be converted to a
CustomGBForceimplementation.CustomGBForceThis will work only of the energy function and parameter names are the same; otherwise, an
Exceptionwill be thrown.Parameters will be interpreted between the two systems.
Exclusions will be combined from both systems.
Exclusions in the shared components of the system must match.
Other forces
Right now, we will throw an
Exception, but it is possible we will want to treat other forces (MonteCarloBarostat,COMMotionRemover) by checking that all parameters are the same and adding it to the resulting alchemically-modified systemAnything I have forgotten?
The text was updated successfully, but these errors were encountered: