{kind=link}
| title | author | date | output | vignette |
|---|---|---|---|---|
Sparse Partial correlation ON Gene Expression with SPONGE |
Markus List, Azim Dehghani Amirabad, Dennis Kostka, Marcel H. Schulz |
`r Sys.Date()` |
rmarkdown::html_vignette |
%\VignetteIndexEntry{SPONGE vignette} %\VignetteEngine{knitr::rmarkdown} %\VignetteEncoding{UTF-8}
|
SPONGE is the first method to solve the computationally demanding task of reporting significant ceRNA interactions efficiently on a genome-wide scale. Beyond ceRNAs, this method is well suited to infer other types of regulatory interactions such as transcription factor regulation.
MicroRNAs (miRNAs) are small 19-22 nucleotide long molecules that facilitate the degradation of messenger RNA (mRNA) transcripts targeted via matching seed sequences. The competing endogenous RNA (ceRNA) hypothesis suggests that mRNAs that possess binding sites for the same miRNAs are in competition. This motivates the existence of so-called sponges, i.e., genes that exert strong regulatory control via miRNA binding in a ceRNA interaction network. It is currently an unsolved problem how to estimate miRNA-mediated ceRNA interactions genome-wide. The most widely used approach considers miRNA and mRNA expression jointly measured for the same cell state. Several partial association methods have been proposed for determining ceRNA interaction strength using conditional mutual information or partial correlation, for instance.
However, we identified three key limitations of existing approaches that prevent the construction of an accurate genome-wide ceRNA interaction network: (i) none of the existing methods considers the combinatorial effect of several miRNAs; (ii) due to the computational demand, the inference of a ceRNA interaction for all putative gene-miRNA-gene interactions in the human genome is prohibitive; (iii) an efficient strategy for determining the significance of inferred ceRNA interactions is missing, and thus important parameters of the system are neglected.
To overcome these challenges, we developed a novel method called Sparse partial correlation on gene expression (SPONGE). We reduce the computational complexity of constructing a genome-wide ceRNA interaction network in several steps. First, we consider only miRNA-gene interactions that are either predicted or experimentally validated. Second, we retain only miRNA-gene interactions that have a negative coefficient in a regularized regression model. Third, instead of each gene-miRNA-gene triplet, we compute a single sensitivity correlation (correlation - partial correlation) for each gene-gene pair given all shared miRNAs that pass the above filter as putative regulators. Finally, we derived the first mathematical formulation to simulate the null distribution of the process for different parameters of the system: number of miRNAs, correlation between genes and sample number. Our formulation enables the computation of empirical p-values for the statistic in a very efficient manner, an order of magnitude faster than previous methods. Analyses revealed that previous studies have underestimated the effect of these parameters in their network inference. Network centrality measures can be applied to SPONGE inferred ceRNA networks to reveal known and novel sponges, many of which are potential biomarkers.
Further details demonstrating how SPONGE improves over the state of the art and how SPONGE inferred ceRNA networks can be used for biomarker discovery will be available in our paper (manuscript submitted, link will be included here at a later point).
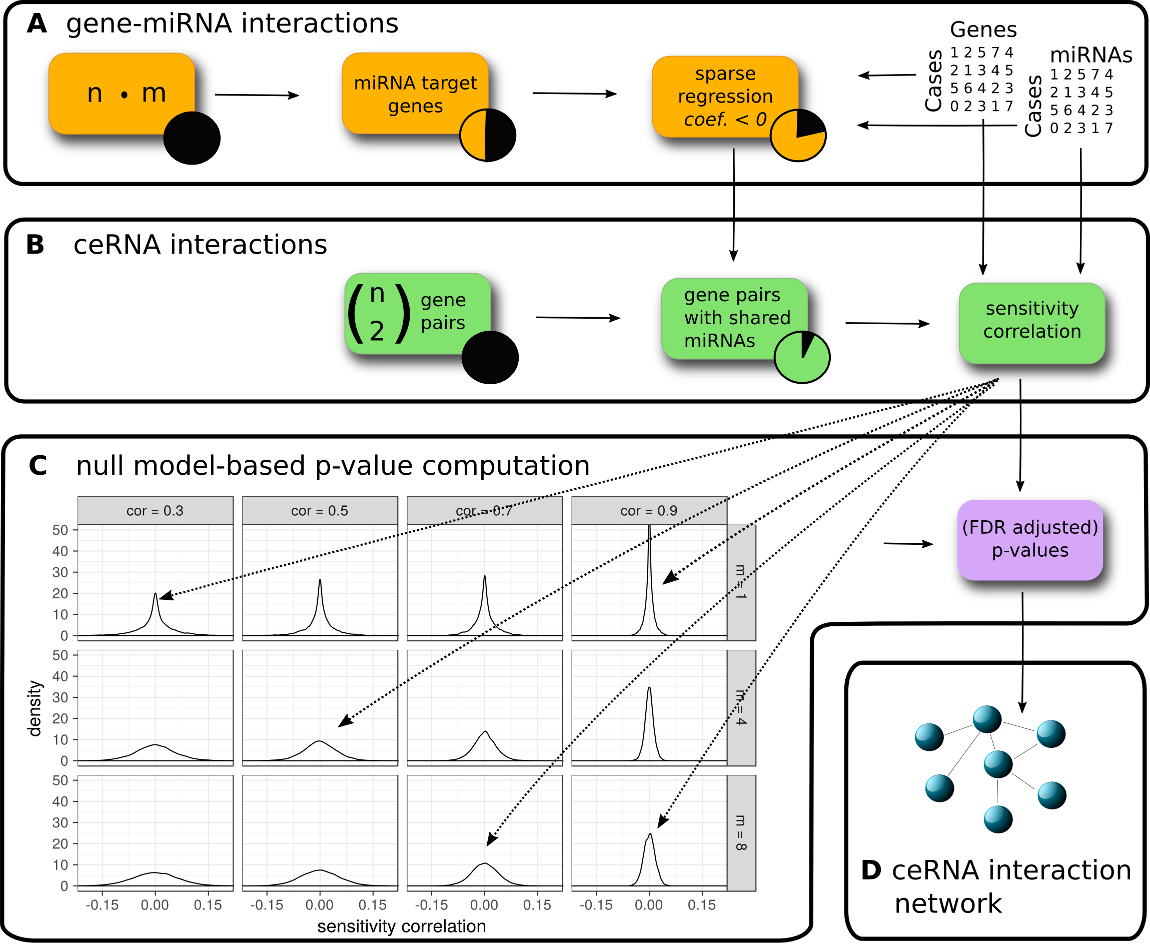
Overview of the SPONGE workflow. (A) Predicted and/or experimentally validated gene-miRNA interactions are subjected to regularized regression on gene and miRNA expression data. Interactions with negative coefficients for miRNA regulators are retained since they indicate miRNA induced inhibition of gene expression. (B) We compute sensitivity correlation coefficients for gene pairs based on shared miRNAs identified in (A). (C) Given the sample number, we compute empirical null models for various gene-gene correlation coefficients (k, columns) and number of miRNAs (m, rows). Sensitivity correlation coefficients are assigned to the best matching null model and a p-value is inferred. (D) After multiple testing correction, significant ceRNA interactions can be used to construct a genome-wide, disease or tissue-specific ceRNA interaction network. In the following hands-on tutorial, we will highlight how each of these steps can be achieved with the SPONGE R package.
We start with loading the package and its dependencies
library(SPONGE)
SPONGE comes with a very small example gene and miRNA expression dataset useful for illustrating functionality. We suggest obtaining larger expression data sets via the GEOquery or TCGAbiolinks R packages, for instance. After loading the package, the example datasets can be accessed:
Gene expression:
head(gene_expr)
knitr::kable(gene_expr[1:5,1:8])
miRNA expression:
head(mir_expr)
knitr::kable(mir_expr[1:5,1:5])
Note that the expected format for both expression matrices is that columns correspond to genes / miRNAs and rows correspond to samples. Please make sure that samples are ordered identically in both matrices and that each sample has an entry in both matrices (paired gene and miRNA expression data).
SPONGE uses a two-tier filtering approach to identify gene-miRNA interactions.
First, the user may incorporate prior knowledge to narrow down the number of gene-miRNA interactions. This could be, for instance, the output of a sequence-based miRNA target prediction software such as TargetScan or miRcode or experimental evidence obtained from databases such as miRTarBase or LncBase. The user is free to use his or her trusted resource(s) and only needs to make sure that the miRNA-gene interactions are formatted as SPONGE expects it:
SPONGE allows for an arbitrary number of miRNA-gene interaction matrices
head(targetscan_symbol)
knitr::kable(targetscan_symbol[1:5,1:5])
Note that the gene and miRNA identifiers in the interaction matrices need to be of the same type and format as the ones used in the expression matrices. Otherwise they can not be matched by SPONGE.
SPONGE combines user-provided matrices as the ones shown above by summing up their entries and subsequently identifies gene-miRNA interaction candidates with non-zero entries. In the second step, we use sparse regularized regression with the gene expression as dependent and miRNA expression as explanatory variables. More specifically, we use elasticnet (via the R package glmnet) to balance lasso and ridge regression. Lasso drives coefficients of miRNAs with negligible influence on gene expression to zero, while ridge regression prevents us from kicking out correlated miRNAs that have similar influence. Elasticnet has two parameters,
We apply step A of the SPONGE workflow as follows:
genes_miRNA_candidates <- sponge_gene_miRNA_interaction_filter(
gene_expr = gene_expr,
mir_expr = mir_expr,
mir_predicted_targets = targetscan_symbol)
The result is a list of genes, where each entry holds a data frame with retained miRNAs and their coefficients (or p-values in case the F-test is used). Note that the mir_predicted_targets parameter may also be NULL. In this case, no prior knowledge is used and all putative miRNA-gene interactions are used in elasticnet regression.
genes_miRNA_candidates[1:2]
In the second step of the SPONGE workflow, we identify ceRNA actions candidates. SPONGE uses the information from (A) and checks for each pair of genes if they share one or more miRNAs with regulatory effect. This reduces the putative number of ceRNA interactions
\begin{eqnarray}\label{formula_scor} mscor(g_1, g_2, M) & = & cor(g_1, g_2) - pcor(g_1, g_2 | M) \end{eqnarray}
where
We apply step B of the SPONGE workflow as follows:
ceRNA_interactions <- sponge(gene_expr = gene_expr,
mir_expr = mir_expr,
mir_interactions = genes_miRNA_candidates)
The result is a data table with both genes (A and B), the number of miRNAs (\emph{df}), gene-gene correlation (\emph{cor}), partial correlation given all miRNAs (\emph{pcor}) and the \emph{mscor} values, i.e.
head(ceRNA_interactions)
knitr::kable(head(ceRNA_interactions))
A theory for the random distribution of \emph{mscor} values did not exist and previous studies using sensitivity correlation either used simple thresholds or random background distributions inferred from random triplets. Simulation experiments show that this approach does not account for the fact that the distribution of \emph{mscor} parameters is strongly affected by gene-gene correlation and the number of samples considred.
This motivated us to develop a strategy for inferring the distribution of \emph{mscor} values under the null hypothesis that miRNAs do not affect the correlation between two genes, i.e.
mscor_null_model <- sponge_build_null_model(number_of_datasets = 100, number_of_samples = nrow(gene_expr))
We can plot the result of these simulations to see how the distribution is affected by gene-gene correlation and number of miRNAs (if ggplot2 is installed):
sponge_plot_simulation_results(mscor_null_model)
Obviously, this is a bit crude since we only samples 100 data points for each of the partitions to keep the compile time of the vignette in check. The default for the number of data sets to be sampled is muche more reasonable with a value of 1e6.
Using the above null models, SPONGE infers empirical p-values for each of the \emph{mscor} coefficients in step (B). This is achieved by first selecting the closest-matching null model, e.g. closest match for gene-gene correlation and number of miRNAs. Within this partition's random \emph{mscor} distribution we now rank the \emph{mscor} value we computed. This rank can then be used to compute an empirical p-value. Note that the precision, i.e. the smallest p-value obtainable is limited by the number of data sets sampled. For example, sampling 100 data sets as we did here means the best p-value we can get is
After computing p-values for all \emph{mscor} coefficients, we apply multiple testing correction for all \emph{mscor} values within a partition, accounting for the fact that null models with low gene-gene correlation will be frequented substantially more often than those with high gene-gene correlation. To infer p-values we do the following:
ceRNA_interactions_sign <- sponge_compute_p_values(sponge_result = ceRNA_interactions,
null_model = mscor_null_model)
The above method adds two additional columns to the ceRNA results, namely
head(ceRNA_interactions_sign)
knitr::kable(head(ceRNA_interactions_sign))
The last step is fairly straight-forward. We decide on an FDR cutoff for selecting significant ceRNA interactions, e.g. FDR < 0.01 and extract those from the result.
ceRNA_interactions_fdr <- ceRNA_interactions_sign[which(ceRNA_interactions_sign$p.adj < 0.2),]
knitr::kable(head(ceRNA_interactions_fdr))
The resulting network can also be plotted
sponge_plot_network(ceRNA_interactions_fdr, genes_miRNA_candidates)
The significant ceRNA interactions from above can be used to construct a ceRNA network in which genes (ceRNAs) are nodes and edges are drawn when a significant ceRNA interaction is predicted. SPONGE constructs such a network using the igraph R package and computed different network statistics for individual genes. Eigenvector and betweenness centrality can be used to assess the importance of a gene in the ceRNA network. In this example we only selected very few genes which makes this exercise a bit futile. However, if applied to larger gene sets, betweenness centrality reveals powerful ceRNAs that may pose biomarkers of miRNA regulation.
network_centralities <- sponge_node_centralities(ceRNA_interactions_fdr)
Note that additional information about the ceRNA effect strength can be incorporated via weighted eigenvector or betweeness centrality. In this case, we add a column weight to our results table as follows:
ceRNA_interactions_fdr_weight <- ceRNA_interactions_fdr
ceRNA_interactions_fdr_weight$weight <- -log10(ceRNA_interactions_fdr$p.adj)
weighted_network_centralities <- sponge_node_centralities(ceRNA_interactions_fdr)
We can also plot these results with a node degree distribution on top and centrality vs node degree below. The parameter top controls how many of the top x samples are labelled.
sponge_plot_network_centralities(weighted_network_centralities, top = 1)
You can also select individual plots
sponge_plot_network_centralities(weighted_network_centralities, measure = "btw", top = 1)
Even though SPONGE is quite fast in inferring ceRNA interactions it will still take a few days to process a dataset with hundred of samples on a single CPU. We thus built SPONGE on top of the foreach R package:
The foreach R package will allow you to parallelize SPONGE over multiple CPUs. The beauty of this approach is that it is up to the user to decide which backend to use for this. We refer to the foreach package for details. One of the most used backends for foreach is the doParallel R package. Once a parallel compute environment is set up, SPONGE can use it automatically (no extra parameters needed):
library(doParallel)
library(foreach)
num.of.cores <- 2 #many more on a compute cluster
#if you want to use logging
#logging.file <- "where_my_log_file_should_go.log"
logging.file <- NULL
cl <- makeCluster(num.of.cores, outfile=logging.file)
registerDoParallel(cl)
genes_miRNA_candidates <- sponge_gene_miRNA_interaction_filter(
gene_expr = gene_expr,
mir_expr = mir_expr,
mir_predicted_targets = targetscan_symbol)
ceRNA_interactions <- sponge(
gene_expr = gene_expr,
mir_expr = mir_expr,
mir_interactions = genes_miRNA_candidates)
stopCluster(cl)
Note that if you would like to use the BiocParallel package you can simply register any parallel foreach backend such as the one used above as follows:
library("BiocParallel")
register(DoparParam(), default = TRUE)
If you have access to a computing cluster, you can register it as parallel backend using the clustermq package. For details refer to the clustermq package.
library(clustermq)
register_dopar_cmq(n_jobs=2)
A frequently encountered caveat of foreach is that each of the parallel workers will require a copy of the data its operating on. With large datasets this becomes an issue even on compute servers with large memory. We thus support the bigmemory R package to allow each parallel worker process / thread to access data in shared memory. To make use of the big memory package simply replace the gene_expr and mir_expr parameters with bigmemory description objects. We refer to the documentation of the bigmemory package for details. If you have access to a compute server with 40-60 cores, you can typically compute a genome-wide ceRNA network within a few hours.
SPONGE allows for sampling covariance matrices given a gene-gene correlation and a specific number of miRNAs. The package contains a number of precomputed matrices for various parameter combinations. However, it is also possibel to compute additional matrices:
more_covariance_matrices <- sample_zero_mscor_cov(m = 1,
number_of_solutions = 10,
gene_gene_correlation = 0.5)
These can be used to simulate data to sample from the random distribution of \emph{mscor} coefficients given the number of samples available in the expression data.
mscor_coefficients <- sample_zero_mscor_data(cov_matrices = more_covariance_matrices,
number_of_samples = 200, number_of_datasets = 100)
hist(unlist(mscor_coefficients), main = "Random distribution of mscor coefficients")
SPONGE was primarily designed to infer ceRNA interactions efficiently. We point out, however, that the principle behind SPONGE for infering partial associations is applicable to other types of regulation in molecular biology. SPONGE is not limited to negative regulation as it is the case with miRNA regulation and can thus also be useful to study, for instance, RNA binding protein regulation or transcription factor regulation. Moreover, we could envision that this approach is also applicable outside of bioinformatics where partial association is of interest. To our knowledge, SPONGE presents the first method to sample random covariance matrices under the null hypothesis that partial correlation and correlation are equal, which may be useful as a starting point to gain a deeper understanding of the theoretical properties of partial correlations in the future.
We inferred ceRNA regulatory networks for 22 cancer types and a pan-cancer ceRNA network based on data from The Cancer Genome Atlas. To make these networks accessible to the biomedical community, we present SPONGEdb, a database offering a user-friendly web interface to browse and visualize ceRNA interactions and an application programming interface accessible by accompanying R and Python packages. SPONGEdb allows researchers to identify potent ceRNA regulators via network centrality measures and to assess their potential as cancer biomarkers through survival, cancer hallmark and gene set enrichment analysis. In summary, SPONGEdb is a feature-rich web resource supporting the community in studying ceRNA regulation within and across cancer types.
SPONGEdb website:
http://sponge.biomedical-big-data.de
API:
http://sponge-api.biomedical-big-data.de
R-package:
https://github.com/biomedbigdata/SPONGE-web-R
Python-package:
https://github.com/biomedbigdata/spongeWebPy
Static-file-server:
http://sponge-files.biomedical-big-data.de
We introduce spongEffects, a novel method that infers functional subnetworks, or modules, and calculates scores related to their activity. We show how spongEffects can be used for downstream machine learning tasks like tumor classification and can identify subtype-specific regulatory interactions that might be further exploited as biomarkers. We show that spongEffects reliably distinguishes between breast cancer subtypes Luminal A, Luminal B, Basal, He+, and Normal-like. Further, we show that spongEffects, once trained on a ceRNA network, can also distinguish between cancer subtypes on datasets and cohorts that do not include miRNA information.
spongEffects is available in this package
To cite your use of SPONGE, SPONGEdb, and/or spongEffects in your publication, please reference one or more of:
List, M., Dehghani Amirabad, A., Kostka, D., & Schulz, M. H. (2019). Large-scale inference of competing endogenous RNA networks with sparse partial correlation. Bioinformatics, 35(14), i596-i604. doi:10.1093/bioinformatics/btz314
Hoffmann, M., Pachl, E., Hartung, M., Stiegler, V., Baumbach, J., Schulz, M. H., & List, M. (2021). SPONGEdb: a pan-cancer resource for competing endogenous RNA interactions. In NAR Cancer (Vol. 3, Issue 1). https://doi.org/10.1093/narcan/zcaa042
Boniolo F., Hoffmann, M., Castro M., Shmulevich I., Tercan B., Roggendorf N., Baumbach J., Robertson G., Saur D., Hennighausen L., & List, M. (2022). spongEffects: patient-specific ceRNA sponging effects in cancer biology to identify subtypes and biomarkers In NAR Cancer (Vol. 4, Issue 2). INSERT DOI ONCE PUBLISHED