PhyloSuite is an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. It combines the functions of two previous tools: MitoTool (https://github.com/dongzhang0725/MitoTool) and BioSuite (https://github.com/dongzhang0725/BioSuite).
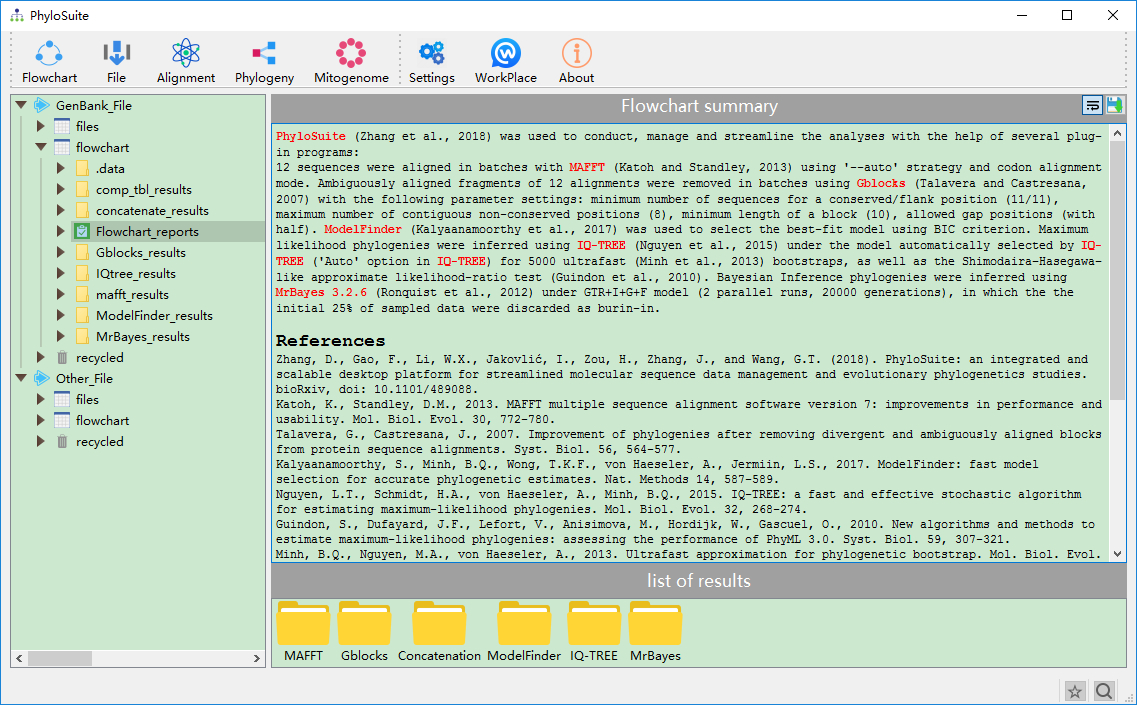
The interface and the main functions of PhyloSuite.
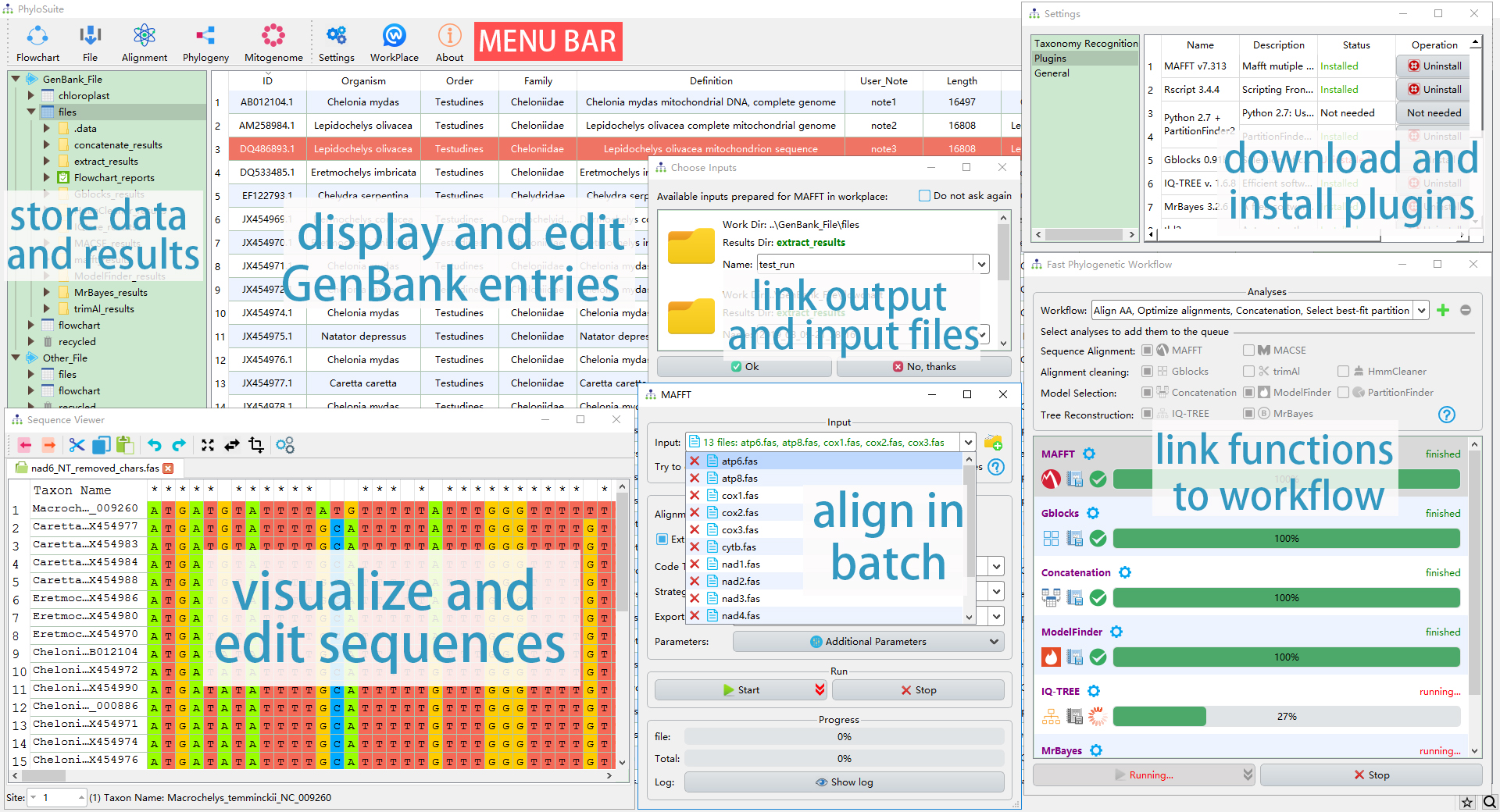
The workflow diagram of PhyloSuite.
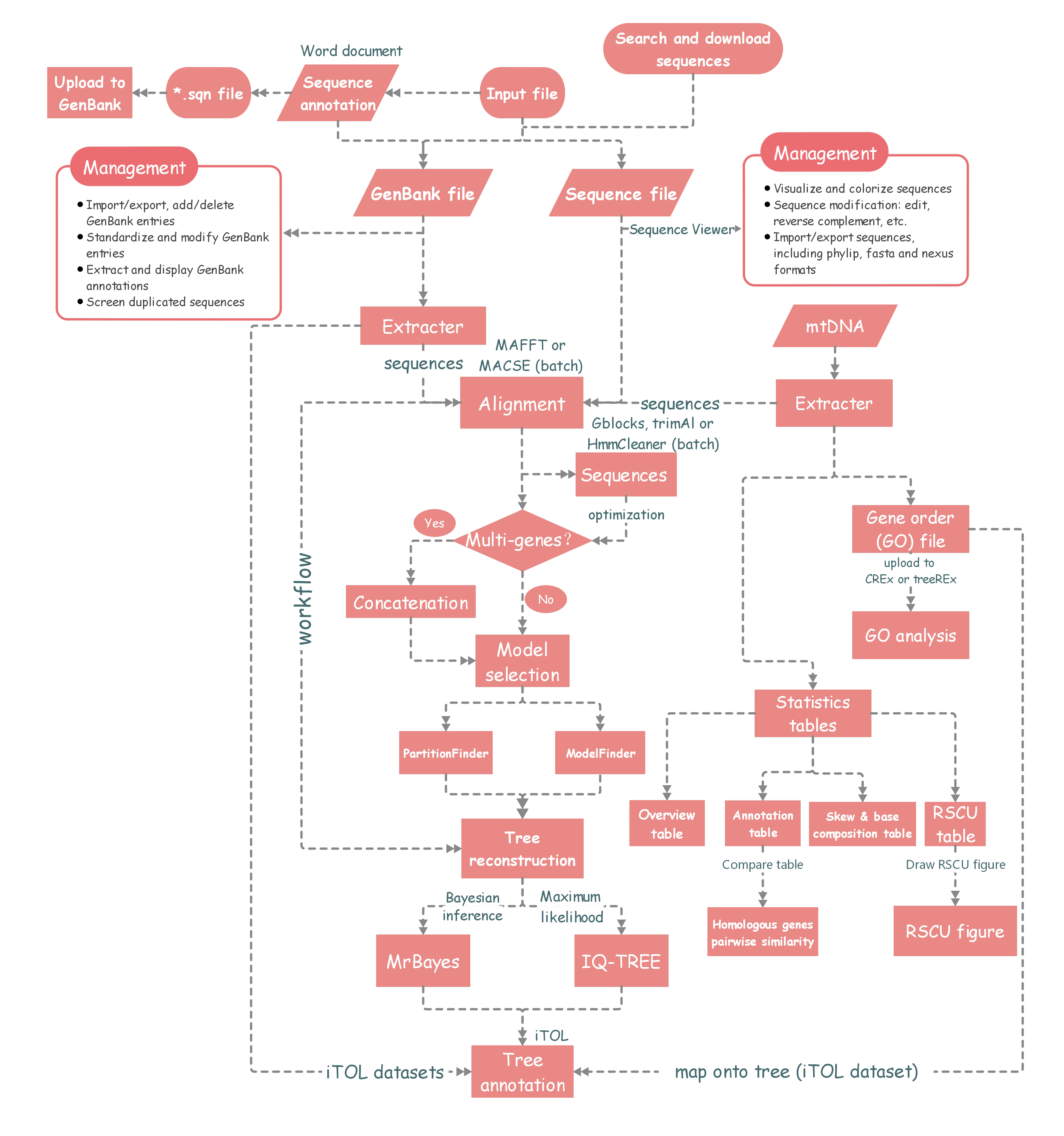
The summary of flowchart and references of used programs.
https://dongzhang0725.github.io or http://phylosuite.jushengwu.com/ (China)
Installers for all platforms can be downloaded from https://github.com/dongzhang0725/PhyloSuite/releases.
Windows 7, 8 and 10 are supported, just double click the PhyloSuite_xxx_win_setup.exe to install, and run “PhyloSuite.exe” file after the installation. If the installation fails, download PhyloSuite_xxx_Win.rar, unzip it, and run PhyloSuite directly from this folder.
Unzip PhyloSuite_xxx_Mac.zip/PhyloSuite_xxx_Linux.tar.gz to anywhere you like, and double click “PhyloSuite” (in PhyloSuite folder) to start, or use the following command:
cd path/to/PhyloSuite
./PhyloSuite
If you encounter an error of "permission denied", try to use the following command:
chmod -R 755 path/to/PhyloSuite(folder)
Note that both 64 bit and 32 bit Windows is supported (Windows 7 and above), whereas only 64 bit has been tested in Linux (Ubuntu 14.04.1 and above) and Mac OSX (macOS Sierra version 10.12.3 and above).
First, Python (version higher than 3.6) should be installed and added to the environment variable in your computer. Then open the terminal and type:
pip install PhyloSuite
It will take some time to install. If it installs successfully, PhyloSuite will be automatically added to the environment variables. Then open the terminal again and type:
PhyloSuite
If the above pip command failed to install PhyloSuite, you can use compiled PhyloSuite (see section 1) or find and download the source codes here (https://pypi.org/project/PhyloSuite/#files or https://github.com/dongzhang0725/PhyloSuite), and install it manually.
Similarly, Python (version higher than 3.6) should be pre-installed and added to the environment variable in your computer. If it is, then download the PhyloSuite from github, either using git clone https://github.com/dongzhang0725/PhyloSuite.git or use the Clone or download-->Download ZIP button in https://github.com/dongzhang0725/PhyloSuite.
After that, change your directory to the PhyloSuite folder that contains the setup.py file, then type:
python setup.py install
https://raw.githubusercontent.com/dongzhang0725/PhyloSuite/master/example.zip
Google group, Github issue or send email to dongzhang0725@gmail.com.
Zhang, D., F. Gao, I. Jakovlić, H. Zou, J. Zhang, W.X. Li, and G.T. Wang, PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular Ecology Resources, 2020. 20(1): p. 348–355. DOI: 10.1111/1755-0998.13096. (Download as: RIS XML ENW)