Annotation
In this tutorial we will use annotations obtained from Ensembl (Homo_sapiens.GRCh38.86.gtf.gz) for chromosome 22 only. For time reasons, these are prepared for you and made available on your AWS instance. But you should get familiar with sources of gene annotations for RNA-seq analysis.
Copy the gene annotation files to the working directory.
echo $RNA_REFS_DIR
cd $RNA_REFS_DIR
wget http://genomedata.org/rnaseq-tutorial/annotations/GRCh38/chr22_with_ERCC92.gtf
Take a look at the contents of the gtf file. Press 'q' to exit the 'less' display.
echo $RNA_REF_GTF
less -p start_codon -S $RNA_REF_GTF
How many unique gene IDs are in the .gtf file?
We can use a perl command-line command to find out:
perl -ne 'if ($_ =~ /(gene_id\s\"ENSG\w+\")/){print "$1\n"}' $RNA_REF_GTF | sort | uniq | wc -l
- Using perl -ne '' will execute the code between single quotes, on the .gtf file, line-by-line.
- The $_ variable holds the contents of each line.
- The 'if ($_ =~//)' is a pattern-matching command which will look for the pattern "gene_id" followed by a space followed by "ENSG" and one or more word characters (indicated by \w+) surrounded by double quotes.
- The pattern to be matched is enclosed in parentheses. This allows us to print it out from the special variable $1.
- The output of this perl command will be a long list of ENSG Ids.
- By piping to sort, then uniq, then word count we can count the unique number of genes in the file.
Now view the structure of a single transcript in GTF format. Press 'q' to exit the 'less' display when you are done.
grep ENST00000342247 $RNA_REF_GTF | less -p "exon\s" -S
To learn more, see:
- http://perldoc.perl.org/perlre.html#Regular-Expressions
- http://www.perl.com/pub/2004/08/09/commandline.html
-
Reference genome - The nucleotide sequence of the chromosomes of a species. Genes are the functional units of a reference genome and gene annotations describe the structure of transcripts expressed from those gene loci.
-
Gene annotations - Descriptions of gene/transcript models for a genome. A transcript model consists of the coordinates of the exons of a transcript on a reference genome. Additional information such as the strand the transcript is generated from, gene name, coding portion of the transcript, alternate transcript start sites, and other information may be provided.
-
GTF (.gtf) file - A common file format referred to as Gene Transfer Format used to store gene and transcript annotation information. You can learn more about this format here: http://genome.ucsc.edu/FAQ/FAQformat#format4
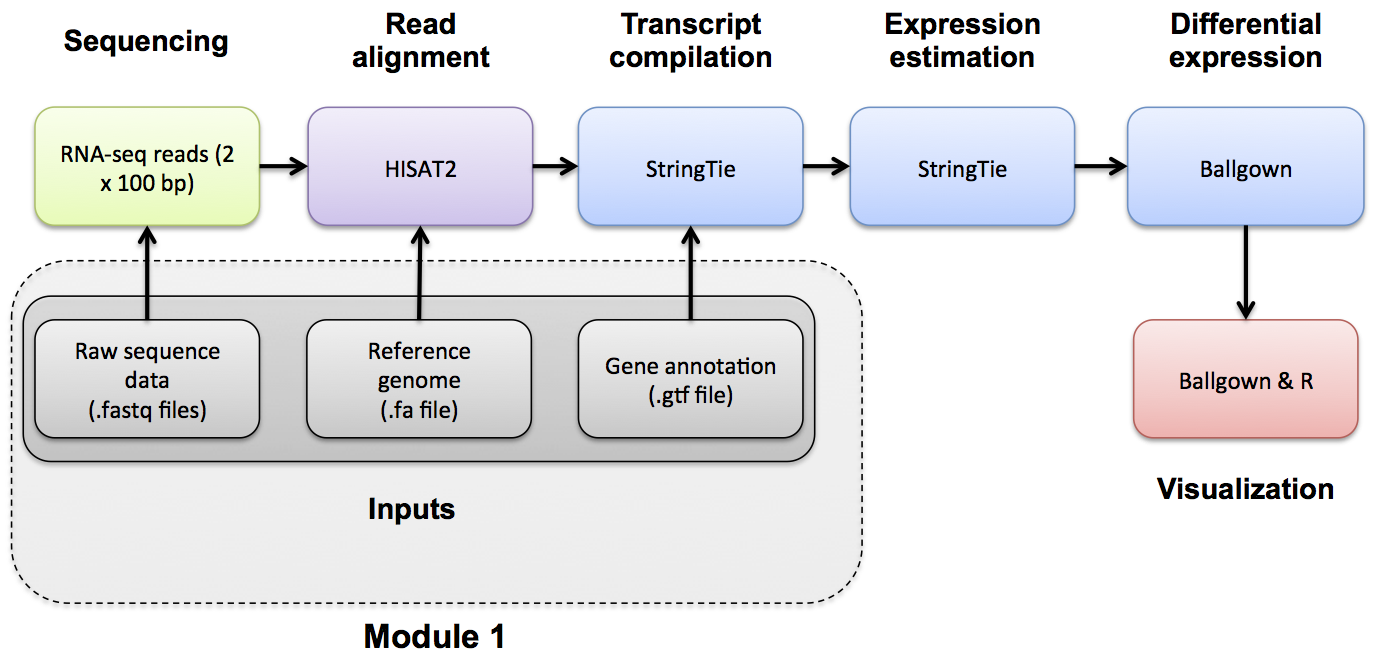
When running the HISAT2/StringTie/Ballgown pipeline, known gene/transcript annotations are used for several purposes:
- During the HISAT2 index creation step, annotations may be provided to create local indexes to represent transcripts as well as a global index for the entire reference genome. This allows for faster mapping and better mapping across exon boundaries and splice sites. If an alignment still can not be found it will attempt to determine if the read corresponds to a novel exon-exon junction. See the Indexing section and the HISAT2 publication for more details.
- During the StringTie step, a .gtf file can be used to specify transcript models to guide the assembly process and limit expression estimates to predefined transcripts using the '-G' and '-e' options together. The '-e' option will give you one expression estimate for each of the transcripts in your .gtf file, giving you a 'microarray like' expression result.
- During the StringTie step, if the '-G' option is specified without the '-e' option the .gtf file is used only to 'guide' the assembly of transcripts. Instead of assuming only the known transcript models are correct, the resulting expression estimates will correspond to both known and novel/predicted transcripts.
- During the StringTie and gffcompare steps, a .gtf file is used to determine the transcripts that will be examined for differential expression using Ballgown. These may be known transcripts that you download from a public source, or a .gtf of transcripts predicted by StringTie from the read data in an earlier step.
There are many possible sources of .gtf gene/transcript annotation files. For example, from Ensembl, UCSC, RefSeq, etc. Several options and related instructions for obtaining the gene annotation files are provided below.
Based on Ensembl annotations only. Available for many species. http://useast.ensembl.org/info/data/ftp/index.html
Based on UCSC annotations or several other possible annotation sources collected by UCSC. You might chose this option if you want to have a lot of flexibility in the annotations you obtain. e.g. to grab only the transcripts from chromosome 22 as in the following example:
- Open the following in your browser: http://genome.ucsc.edu/
- Select 'Tools' and then 'Table Browser' at the top of the page.
- Select 'Mammal', 'Human', and 'Dec. 2013 (GRCh38/hg38)' from the first row of drop down menus.
- Select 'Genes and Gene Predictions' and 'GENCODE v24' from the second row of drop down menus. To limit your selection to only chromosome 22, select the 'position' option beside 'region', enter 'chr22' in the 'position' box.
- Select 'GTF - gene transfer format' for output format and enter 'UCSC_Genes.gtf' for output file.
- Hit the 'get output' button and save the file. Make note of its location
In addition to the .gtf file you may find uses for some extra files providing alternatively formatted or additional information on the same transcripts. For example: How to get a Gene bed file:
- Change the output format to 'BED - browser extensible data'.
- Change the output file to 'UCSC_Genes.bed', and hit the 'get output' button.
- Make sure 'Whole Gene' is selected, hit the 'get BED' button, and save the file.
How to get an Exon bed file:
- Go back one page in your browser and change the output file to 'UCSC_Exons.bed', then hit the 'get output' button again.
- Select 'Exons plus', enter 0 in the adjacent box, hit the 'get BED' button, and save the file.
How to get gene symbols and descriptions for all UCSC genes:
- Again go back one page in your browser and change the 'output format' to 'selected fields from primary and related tables'.
- Change the output file to 'UCSC_Names.txt', and hit the 'get output' button.
- Make sure 'chrom' is selected near the top of the page.
- Under 'Linked Tables' make sure 'kgXref' is selected, and then hit 'Allow Selection From Checked Tables'. This will link the table and give you access to its fields.
- Under 'hg38.kgXref fields' select: 'kgID', 'geneSymbol', 'description'.
- Hit the 'get output' button and save the file.
To get annotations for the whole genome, make sure 'genome' is selected beside 'region'. By default, the files downloaded above will be compressed. To decompress, use 'gunzip filename' in linux.
HISAT2 has prebuilt reference genome index files for both DNA and RNA alignment. Various versions of the index files include SNPs and/or transcript splice sites. Versions of both the Ensembl and UCSC genomes for human build 38 are linked from the main HISAT2 page: https://ccb.jhu.edu/software/hisat2/index.shtml
Or those same files are directly available from their FTP site: ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/
On chromosome naming conventions:
In order for your RNA-seq analysis to work, the chromosome names in your .gtf file must match those in your reference genome (i.e. your reference genome fasta file). If you get a StringTie result where all transcripts have an expression value of 0, you may have overlooked this. Unfortunately, Ensembl, NCBI, and UCSC can not agree on how to name the chromosomes in many species, so this problem may come up often. You can avoid this by getting a complete reference genome and gene annotation package from the same source (e.g., Ensembl) to maintain consistency.
On reference genome builds:
Your annotations must correspond to the same reference genome build as your reference genome fasta file. e.g., both correspond to UCSC human build 'hg38', NCBI human build 'GRCh38', etc. Even if both your reference genome and annotations are from UCSC or Ensembl they could still correspond to different versions of that genome. This would cause problems in any RNA-seq pipeline.
| Previous Section | This Section | Next Section |
|---|---|---|
| Reference Genomes | Annotations | Indexing |