calculateFragments() missing features #47
Comments
|
I am not quite sure that I understand your feature request completely:
Currently
You are right. In the current implementation the highest matched peak is reported. I will think about a way to report all peaks in the user-defined tolerance range.
Correct. Would it be enough to just substract the mass of NH3 respectively H2O? Or do we need a more sophisticated rule? |
|
Sorry if things were not clear enough 3| I think a substraction of respectively 17 and 18 is sufficient. H20-NH3 is also fairly common. I found a table with some common neutral losses online. https://books.google.be/books?id=34ErvND2KLwC&pg=PA16&lpg=PA16&dq=proteomics+H2O+nh3+loss&source=bl&ots=Uf4SFLuW9K&sig=KvYL1nwLxtyzj9JF_J0sCs8ZEZw&hl=en&sa=X&ei=7jbPVOClOJHwaOTdgJgF&ved=0CC8Q6AEwAg#v=onepage&q=proteomics%20H2O%20nh3%20loss&f=false |
|
regarding 3. Thanks for the table! Should we report neutral losses for all possible fragments? According to http://www.matrixscience.com/help/fragmentation_help.html Mascot reports loss of ammonia or water only for fragments that contain |
|
Hi Well I did a small google search to track down the origin of this 'rule' but all I can find are references to this Mascot document and no original source. So I'm not sure if this holds in all cases. For example following document seems like a thorough review of fragmentation pathways and they never mention this. (http://cbio.ufs.ac.za/fgap/download/fragmentation_review.pdf ) Maybe safest thing would be to allow everything and let the user decide? (No I think of it. Immonium ions would maybe also be a nice addition to complete the functionality of the fragmentation tool, if it's not too much work off course) |
|
Thanks for share this great review. I just scanned the document but it seems to support the Mascot "rule": water loss:
The mentioned C-terminal COOH group means that nearly every peptide can be affected by water loss (so maybe we should not overcomplicate our implementation and report just every possibility). ammonia loss:
On page 4 there is a great overview: |

|
It's hard to see for me how they can loose water at backbone amide oxygen atoms. They provide an example of all cases in this paper except of that. Maybe they mean through interaction with the -COOH termini. (of which they provide an example) If that's the case then there is only water loss at fragments with -COOH (x,y,z) |
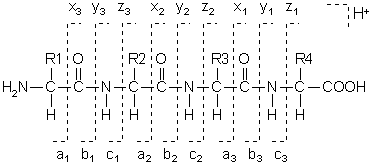
|
I add a basic prototype of a neutral loss function. It is intended as post-processing to .neutralLoss(.calculateFragments("LAAGKVEDSD"))
# mz ion type pos z seq
# 1 114.0913 b1 b 1 1 L
# 2 185.1284 b2 b 2 1 LA
# 3 256.1656 b3 b 3 1 LAA
# 4 313.1870 b4 b 4 1 LAAG
# 5 441.2820 b5 b 5 1 LAAGK
# 6 540.3504 b6 b 6 1 LAAGKV
# 7 669.3930 b7 b 7 1 LAAGKVE
# 8 784.4199 b8 b 8 1 LAAGKVED
# 9 871.4519 b9 b 9 1 LAAGKVEDS
# 10 986.4789 b10 b 10 1 LAAGKVEDSD
# 11 134.0448 y1 y 1 1 D
# 12 221.0768 y2 y 2 1 SD
# 13 336.1038 y3 y 3 1 DSD
# 14 465.1463 y4 y 4 1 EDSD
# 15 564.2148 y5 y 5 1 VEDSD
# 16 692.3097 y6 y 6 1 KVEDSD
# 17 749.3312 y7 y 7 1 GKVEDSD
# 18 820.3683 y8 y 8 1 AGKVEDSD
# 19 891.4054 y9 y 9 1 AAGKVEDSD
# 20 1004.4895 y10 y 10 1 LAAGKVEDSD
# 21 101.0471 y1_ y_ 1 1 D
# 22 188.0792 y2_ y_ 2 1 SD
# 23 303.1061 y3_ y_ 3 1 DSD
# 24 432.1487 y4_ y_ 4 1 EDSD
# 25 531.2171 y5_ y_ 5 1 VEDSD
# 26 659.3121 y6_ y_ 6 1 KVEDSD
# 27 716.3335 y7_ y_ 7 1 GKVEDSD
# 28 787.3706 y8_ y_ 8 1 AGKVEDSD
# 29 858.4077 y9_ y_ 9 1 AAGKVEDSD
# 30 971.4918 y10_ y_ 10 1 LAAGKVEDSD
# 31 953.4812 b10_ b_ 10 1 LAAGKVEDSD
# 32 523.3238 b6* b* 6 1 LAAGKV
# 33 652.3664 b7* b* 7 1 LAAGKVE
# 34 767.3934 b8* b* 8 1 LAAGKVED
# 35 854.4254 b9* b* 9 1 LAAGKVEDS
# 36 969.4523 b10* b* 10 1 LAAGKVEDSD
# 37 675.2832 y6* y* 6 1 KVEDSD
# 38 732.3046 y7* y* 7 1 GKVEDSD
# 39 803.3417 y8* y* 8 1 AGKVEDSD
# 40 874.3788 y9* y* 9 1 AAGKVEDSD
# 41 987.4629 y10* y* 10 1 LAAGKVEDSDDo you have any suggestions? |
|
Nice work! Is there is is reason why you implement it as a post processing tool? |
|
accidentely closed |
Because it depends on the results of
I am going to add an argument |
|
## create basic MSnExp
file <- dir(system.file(package = "MSnbase", dir = "extdata"),
full.name = TRUE, pattern = "mzXML$")
msexp <- readMSData(file)
## centroid them
msexp <- pickPeaks(msexp)
calculateFragments("VESITARHGEVLQLRPK", msexp[[1]])
# Modifications used: C=160.030649
# mz intensity ion type pos z seq error
# 1 114.1109 706555.7 y1_ y_ 1 1 K 0.00425275
# 2 429.2563 1972344.0 b4 b 4 1 VESI -0.02189010
# 3 513.3047 2574137.0 y4 y 4 1 LRPK 0.04598246
# 4 754.4504 537234.8 y6 y 6 1 LQLRPK 0.04293155
# 5 982.5354 500159.1 y8 y 8 1 EVLQLRPK 0.06897061
# 6 1080.5867 209363.7 b10 b 10 1 VESITARHGE -0.04344392
# 7 1688.0375 136748.8 y15* y* 15 1 SITARHGEVLQLRPK -0.07729359
# 8 1882.0074 149649.1 b17_ b_ 17 1 VESITARHGEVLQLRPK 0.08206471
calculateFragments("VESITARHGEVLQLRPK", msexp[[1]], neutralLoss=FALSE)
# Modifications used: C=160.030649
# mz intensity ion type pos z seq error
# 1 429.2563 1972344.0 b4 b 4 1 VESI -0.02189010
# 2 513.3047 2574137.0 y4 y 4 1 LRPK 0.04598246
# 3 754.4504 537234.8 y6 y 6 1 LQLRPK 0.04293155
# 4 982.5354 500159.1 y8 y 8 1 EVLQLRPK 0.06897061
# 5 1080.5867 209363.7 b10 b 10 1 VESITARHGE -0.04344392 |
|
Great, I'll try to test it out as soon as possible. thanks for the effort |
|
How do we define a modification at N-terminal? eg. TMT or iTRAQ plex always add a mass=229.1629 to the amino group at N-terminal. Please help me, I am desperate now. |
|
The I am not very familiar with TMT or iTRAQ but wouldn't it be enough to add 229.1629 to your a, b, c fragments? fragments <- calculateFragments("AA", type=c("a", "b", "c", "x", "y", "z"))
abc <- grepl("[abc]", fragments$type)
fragments$mz[abc] <- fragments$mz[abc] + 229.1629
fragments
# mz ion type pos z seq
# 1 273.21237 a1 a 1 1 A
# 2 344.24948 a2 a 2 1 AA
# 3 301.20729 b1 b 1 1 A
# 4 372.24440 b2 b 2 1 AA
# 5 318.23383 c1 c 1 1 A
# 6 389.27094 c2 c 2 1 AA
# 7 116.03422 x1 x 1 1 A
# 8 187.07133 x2 x 2 1 AA
# 9 90.05495 y1 y 1 1 A
# 10 161.09206 y2 y 2 1 AA
# 11 73.02840 z1 z 1 1 A
# 12 144.06551 z2 z 2 1 AA
# 13 83.03656 x1_ x_ 1 1 A
# 14 154.07367 x2_ x_ 2 1 AA
# 15 57.05730 y1_ y_ 1 1 A
# 16 128.09441 y2_ y_ 2 1 AA
# 17 40.03075 z1_ z_ 1 1 A
# 18 111.06786 z2_ z_ 2 1 AA |
|
@sgibb , thanks for the answer! Because I want to use "calculateFragments" method to compare the fragment ions of a peptide sequence against a spectrum object, I need to add the modifications in the method, such as. calculateFragments("VESITARHGEVLQLRPK", msexp[[1]], modifications=c(N-term ions + 229.1629)). Is it possible to do like this now? |
|
@lgatto: How do you handle reporter ions? Would it be worth to add a |
|
Not sure if |
|
I would prefer adding
|
|
regarding 2: IMHO using |
Yes. The this first figures here Mascot site.
Yes, I think this is a good suggestion, and would make the interface consistent with new feature. This is however a major change in the interface, so a message explaining this whenever the function is called (that we would keep during a full release cycle) would seem appropriate. Not sure if we want to also keep the old behaviours optional.
I like your suggestions. |
Before the modification listed in "modifications" was used to replace the mass of an amino acid. Now it is added to the mass. See #47 for details.
|
The mass in calculateFragments("AA")
# The mass listed in "modifications" is now added to the amino acid/peptide.
# In MSnbase < 1.17.6 the mass was replaced. Please see '?calculateFragments' for details.
# mz ion type pos z seq
# 1 72.04439 b1 b 1 1 A
# 2 143.08150 b2 b 2 1 AA
# 3 90.05495 y1 y 1 1 A
# 4 161.09206 y2 y 2 1 AA
# 5 57.05730 y1_ y_ 1 1 A
# 6 128.09441 y2_ y_ 2 1 AA
calculateFragments("AA", modifications=c(Nterm=100, Cterm=-90))
# The mass listed in "modifications" is now added to the amino acid/peptide.
# In MSnbase < 1.17.6 the mass was replaced. Please see '?calculateFragments' for details.
# mz ion type pos z seq
# 1 172.044386 b1 b 1 1 A
# 2 243.081496 b2 b 2 1 AA
# 3 0.054951 y1 y 1 1 A
# 4 71.092061 y2 y 2 1 AA
# 5 57.057296 y1_ y_ 1 1 A
# 6 128.094406 y2_ y_ 2 1 AA
calculateFragments("VESITARHGEVLQLRPK", msexp[[1]],
type="a")
# The mass listed in "modifications" is now added to the amino acid/peptide.
# In MSnbase < 1.17.6 the mass was replaced. Please see '?calculateFragments' for details.
# mz intensity ion type pos z seq error
# 1 540.3018 704495.9 a6_ a_ 6 1 VESITA 0.02476715
# 2 1375.7462 213299.7 a13* a* 13 1 VESITARHGEVLQ -0.01340382
calculateFragments("VESITARHGEVLQLRPK", msexp[[1]],
type="a", modifications=c(Nterm=229))
# The mass listed in "modifications" is now added to the amino acid/peptide.
# In MSnbase < 1.17.6 the mass was replaced. Please see '?calculateFragments' for details.
# mz intensity ion type pos z seq error
# 1 1375.746 213299.7 a13* a* 13 1 VESITARHGEVLQ -0.01340382The API change info message is displayed for |
|
@sgibb @lgatto , Great! Thanks for the prompt response! There is an issue I would like to discuss here. As I understand, if you set Nterm=229, the new "calculateFragments" method adds the mass 229 to a,b,c ions only. However, the mass of neutral loss ions are also changed because the Nterm or Cterm modifications, so the neutral loss N-terminal ions ( One more thing regarding modification on K (Lysine). Since TMT or iTRAQ tag will modify both N-term and K through NH2 group, so lysine related ammonia loss won't happen after it is been modified by TMT. For example, calculateFragments("AKA",modifications=c(Nterm=229, K=229)) should not generate any ammonia loss ions. At last, I would really appreciate if you add one option to enable/disable water loss or ammonia loss separately. Sometimes I just want to have water loss ions but not ammonia loss. (Because water loss is much more common to observe than ammonia loss). |
|
@yafeng I was totally blockhead. I somehow thought the neutral loss would affect the Regarding your second issue: It would be no problem to separately enable/disable water/ammonia loss. I will implement it. But the K-problem is somehow more complicated. By modifying K using |
|
@sgibb that is perfect, Thanks! |
|
I just rewrote the underlying .calculateFragments <- function(sequence, type=c("b", "y"), z=1,
modifications=c(C=57.02146),
neutralLoss=
list(water=c("Cterm", "D", "E", "S", "T"),
ammonia=c("K", "N", "Q", "R")),
verbose=TRUE) {Calling the default function would be easy (full neutral loss support): .calculateFragments("AKA", modifications=c(Nterm=229, K=229))
# mz ion type pos z seq
# 1 301.04439 b1 b 1 1 A
# 2 658.13935 b2 b 2 1 AK
# 3 729.17646 b3 b 3 1 AKA
# 4 90.05495 y1 y 1 1 A
# 5 447.14991 y2 y 2 1 KA
# 6 518.18702 y3 y 3 1 AKA
# 7 57.05730 y1_ y_ 1 1 A
# 8 414.15226 y2_ y_ 2 1 KA
# 9 485.18937 y3_ y_ 3 1 AKA
# 10 712.14991 b3* b* 3 1 AKA
# 11 430.12336 y2* y* 2 1 KA
# 12 501.16047 y3* y* 3 1 AKAIf you want to disable ammonia loss just for .calculateFragments("AKA", modifications=c(Nterm=229, K=229),
neutralLoss=list(water=c("Cterm", "D", "E", "S", "T"),
ammonia=c("N", "Q", "R")))
# mz ion type pos z seq
# 1 301.04439 b1 b 1 1 A
# 2 658.13935 b2 b 2 1 AK
# 3 729.17646 b3 b 3 1 AKA
# 4 90.05495 y1 y 1 1 A
# 5 447.14991 y2 y 2 1 KA
# 6 518.18702 y3 y 3 1 AKA
# 7 57.05730 y1_ y_ 1 1 A
# 8 414.15226 y2_ y_ 2 1 KA
# 9 485.18937 y3_ y_ 3 1 AKAI don't like negative arguments like @yafeng , @lgatto Do you have a better idea for the interface? |
|
What about a function or built-in list for the user to easily get the default parameters > x <- defaultNeutralLosses() ## or
> x <- defaultNeutralLosses
> x
$water
[1] "Cterm" "D" "E" "S" "T"
$ammonia
[1] "K" "N" "Q" "R"The function would be something like if (missing(neutralLoss))
neutralLoss <- defaultNeutralLoss()and the user could relatively easily choose the relevant elemets list(x[[1]][-(2:3)], x[[2]][c(1, 4)])
[[1]]
[1] "Cterm" "S" "T"
[[2]]
[1] "K" "R" |
|
@sgibb, I am very satisfied that all my wills are fulfilled now :). Thanks! As for nicer user experience, I agree with @lgatto, it might be a bit difficult for users to list out all other amino acids with ammonia loss. Sometimes they just know the ones they are familiar, for example, I was only aware of ammonia loss on K and R before I see the full list. So if the negative list is not feasible, maybe a built-in list for neutralLoss to make it easier for users to choose. |
|
@lgatto this looks awful 😉
I combined both of your suggestions. There is a new defaultNeutralLoss()
# $water
# [1] "Cterm" "D" "E" "S" "T"
#
# $ammonia
# [1] "K" "N" "Q" "R"
#
defaultNeutralLoss(disableWaterLoss="Cterm", disableAmmoniaLoss=c("K", "R"))
# $water
# [1] "D" "E" "S" "T"
#
# $ammonia
# [1] "N" "Q"
#
## neutral loss activated (internally: neutralLoss=defaultNeutralLoss)
calculateFragments("PQR")
# mz ion type pos z seq
# 1 98.06004 b1 b 1 1 P
# 2 226.11862 b2 b 2 1 PQ
# 3 382.21973 b3 b 3 1 PQR
# 4 175.11895 y1 y 1 1 R
# 5 303.17753 y2 y 2 1 QR
# 6 400.23029 y3 y 3 1 PQR
# 7 142.12130 y1_ y_ 1 1 R
# 8 270.17988 y2_ y_ 2 1 QR
# 9 367.23264 y3_ y_ 3 1 PQR
# 10 365.19318 b3* b* 3 1 PQR
# 11 286.15098 y2* y* 2 1 QR
# 12 383.20374 y3* y* 3 1 PQR
calculateFragments("PQR",
neutralLoss=defaultNeutralLoss(disableWaterLoss="Cterm"))
# mz ion type pos z seq
# 1 98.06004 b1 b 1 1 P
# 2 226.11862 b2 b 2 1 PQ
# 3 382.21973 b3 b 3 1 PQR
# 4 175.11895 y1 y 1 1 R
# 5 303.17753 y2 y 2 1 QR
# 6 400.23029 y3 y 3 1 PQR
# 7 365.19318 b3* b* 3 1 PQR
# 8 286.15098 y2* y* 2 1 QR
# 9 383.20374 y3* y* 3 1 PQR
calculateFragments("PQR",
neutralLoss=defaultNeutralLoss(disableAmmoniaLoss="Q"))
# mz ion type pos z seq
# 1 98.06004 b1 b 1 1 P
# 2 226.11862 b2 b 2 1 PQ
# 3 382.21973 b3 b 3 1 PQR
# 4 175.11895 y1 y 1 1 R
# 5 303.17753 y2 y 2 1 QR
# 6 400.23029 y3 y 3 1 PQR
# 7 142.12130 y1_ y_ 1 1 R
# 8 270.17988 y2_ y_ 2 1 QR
# 9 367.23264 y3_ y_ 3 1 PQR
## disable neutral loss completely
calculateFragments("PQR", neutralLoss=NULL)
# mz ion type pos z seq
# 1 98.06004 b1 b 1 1 P
# 2 226.11862 b2 b 2 1 PQ
# 3 382.21973 b3 b 3 1 PQR
# 4 175.11895 y1 y 1 1 R
# 5 303.17753 y2 y 2 1 QR
# 6 400.23029 y3 y 3 1 PQR@lgatto and @yafeng if you are satified we can merge the |
|
Fine by me. |
|
@sgibb , nice and neat! |
|
I have a problem with modifications. Could you please tell how many maximum modifications allowed for the modification parameter here? I put it like this: modifications=c(C=57.02146, Nterm=144.102063, K=144.102063) and it's not working fine. |
|
@smv1d14 Are you using the latest version? What is the output of |
|
@smv1d14 Sorry, somehow I overlooked this reopened issue. There is no limitation in number of modifications (at least no technical one). Unfortunately I can't reproduce your problem. Using the latest development version of calculateFragments("AKC", modifications=c(C=57.02146, Nterm=144.102063, K=144.102063))
# mz ion type pos z seq
# 1 216.1464 b1 b 1 1 A
# 2 488.3435 b2 b 2 1 AK
# 3 648.3741 b3 b 3 1 AKC
# 4 179.0485 y1 y 1 1 C
# 5 451.2455 y2 y 2 1 KC
# 6 522.2826 y3 y 3 1 AKC
# 7 146.0508 y1_ y_ 1 1 C
# 8 418.2479 y2_ y_ 2 1 KC
# 9 489.2850 y3_ y_ 3 1 AKC
# 10 631.3476 b3* b* 3 1 AKC
# 11 434.2190 y2* y* 2 1 KC
# 12 505.2561 y3* y* 3 1 AKCAs @lgatto already mentioned we need more information (namely a minimal working example that reproduces the error, the output you expected and the output of I will close this issue because it works for me and because lack of information. |
Hi,
I was using the calculateFragments() function in MSnbase and I think there are some small modifications that could make it more usefull.
Have a nice evening
The text was updated successfully, but these errors were encountered: