HowTo: fasterq dump
The fasterq-dump tool extracts data in FASTQ- or FASTA-format from SRA-accessions. It is a commandline-tool that is available for Linux, macOS, and Windows.
It is a part of the SRA-toolkit:
03.-Quick-Toolkit-Configuration
Fasterq-dump is the successor to the older fastq-dump tool, but faster. However: it is not a drop-in replacement, options and defaults are different.
The tool has one mandatory argument: the accession.
example: $fasterq-dump SRR000001
An accession can be specified in 2 different ways:
(1) bare accession: $fasterq-dump SRR000001
or
(2) a file-system path: $fasterq-dump ~/data/SRR000001
For a bare accession, a working internet connection is needed.
For a file-system path, the path can be absolute or relative.
absolute: $fasterq-dump /home/user/data/SRR000001
relative: $fasterq-dump ./SRR000001
An accession is not a file, it is a container of files. Depending in the data submitted to NCBI, the container will have just one file or many files. Because of that, best practice is to always specify a directory.
example: $fasterq-dump /home/user/data/SRR000001
incorrect: $fasterq-dump /home/user/data/SRR000001/SRR000001.sra
The incorrect example may work for some accessions and fail for others.
For best performance it is recommended to use 'prefetch' to download the accession: https://github.com/ncbi/sra-tools/wiki/08.-prefetch-and-fasterq-dump
Fasterq-dump has many options, you can display them by running:
$fasterq-dump -h

If a minimal commandline is given:
$fasterq-dump SRR000001
The tool produces output files named 'SRR000001.fastq', 'SRR000001_1.fastq' and 'SRR000001_2.fastq' in the current directory. The tool will also create a directory named 'fasterq.tmp.host.procid' in the current directory. The host- and procid-parts will be replaced by the hostname of the computer you are using and the process-id. After the extraction is finished, this directory and its content will be deleted. This temporary directory will use approximately up to 10 times the size of the final output-file. If you do not have enough space in your current directory for the output-file and the temporary files, the tool will fail. However it will not always require 10 times the size of the final output-file, this is the worst case. How much space will be required depends on the requested mode. Also more temporary space is required if you request a bare assession instead of a local copy of the accession. In the 'fasta-unsorted'-mode there is no temporary space required, if you use a local copy of the accession.
Fasterq-dump can operate in different modes:

and

The location (output directory) of the output-files can be changed:
$fasterq-dump SRR000001 -O /mnt/big_hddq
If parts of the output-path do not exist, it will be created. If the output-files already exist, the tool will not overwrite them, but fail instead. If you want already existing output-files to be overwritten, use the force option -f.
The location of the temporary directory can be changed too:
$fasterq-dump SRR000001 -O /mnt/big_hdd -t /tmp/scratch
Now the temporary files will be created in the '/tmp/scratch' directory. These temporary files will be deleted on finish, but the directory itself will not be deleted. If the temporary directory does not exist, it will be created.
It is helpful for the speed-up, if the output-path and the scratch-path are on different file-systems. For instance it is a good idea to point the temporary directory to a SSD if available or a RAM-disk like /dev/shm if enough RAM is available.
Another factor is the number of threads. If no option is given (as above) the tool uses 6 threads for its work. If you have more CPU cores it might help to increase this number. The option to do this is -e, for instance -e 8 increases the thread-count to 8. However even if you have a computer with much more CPU cores, increasing the thread count can lead to diminishing returns, because you exhaust I/O - bandwidth. If you specify a bare accession, there might be no gain in speed.
You can test your speed by measuring how long it takes to convert a smaller accession, like this:
$time fasterq-dump data/SRR000001 -t /dev/shm
$time fasterq-dump data/SRR000001 -t /dev/shm -e 8
$time fasterq-dump data/SRR000001 -t /dev/shm -e 10
Don't forget to repeat the commands at least 2 times, to exclude other influences like caching or network load.
To detect how many cpu-cores your machine has:
on Linux: $nproc --all
on Mac: $/usr/sbin/sysctl -n hw.ncpu
The tool can create different formats:
The spots are split into ( biological ) reads, for each read : 4 lines of FASTQ or 2 lines of FASTA are written. For spots having 2 reads, the reads are written into the *_1.fastq and *_2.fastq files. Unmated reads are placed in *.fastq. If the accession has no spots with one single read, the *.fastq-file will not be created.
`example: $fasterq-dump SRR000001`

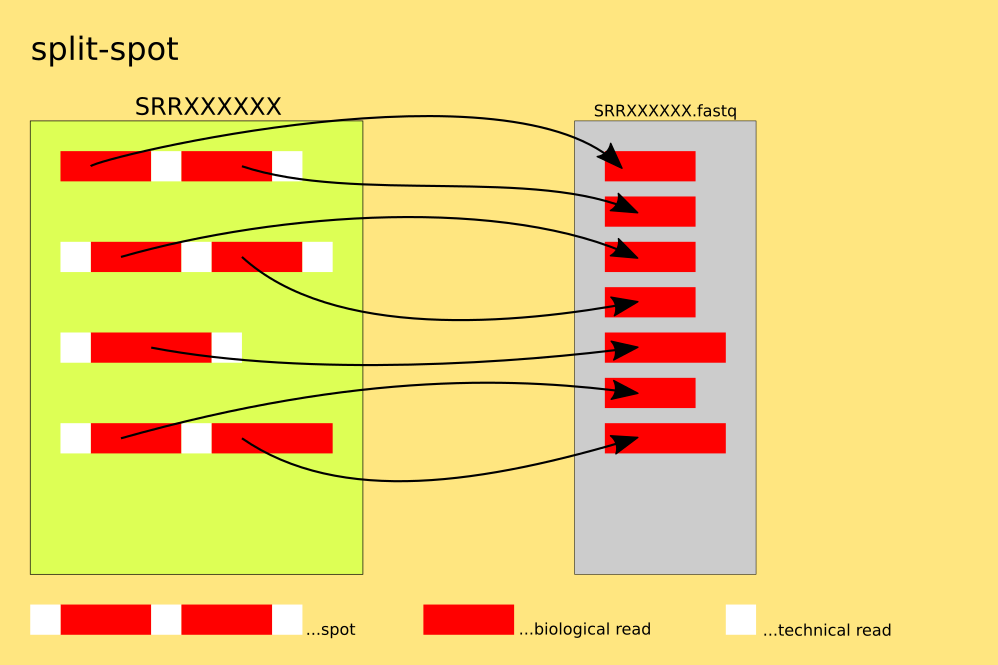
The spots are split into reads, for each read : 4 lines of FASTQ or 2 lines of FASTA are written into the single output-file. This mode allows for the output to be redirected to stdout via: '--stdout ( -Z )'.
`example: $fasterq-dump SRR000001 --split-spot`

The spots are split into reads, for each read : 4 lines of FASTQ or 2 lines of FASTA are written, each n-th read into a different file.
`example: $fasterq-dump SRR000001 --split-file`


The spots are not split : 4 lines of FASTQ or 2 lines of FASTA are written into one output-file for each spot. This mode allows for the output to be redirected to stdout via: '--stdout ( -Z )'.
`example: $fasterq-dump SRR000001 --concatenate-reads`


The spots are split into reads, for each read : 2 lines of FASTA are written into the single output-file. This mode allows for the output to be redirected to stdout via: '--stdout ( -Z )'. This mode is indentical to the split-spot-mode, with the only difference beeing that the original order of the spots and reads is not preserved and it being exlusivly for FASTA. The reason for the existence of this mode is the fact that this mode is faster then the split-spot-mode, and does not use temporary files.
`example: $fasterq-dump SRR000001 --fasta-unsorted`
It is possible that you exhaust the space at your filesystem while converting large accessions. This can happen with this tool more often because it uses additional scratch-space to increase speed. It is a good idea to perform some simple checks before you perform the conversion. First you should know how big an accession is. Let us use the accession SRR341578 as an example:
$vdb-dump --info SRR341578
will give you a lot of information about this accession. The important line is the 3rd one: 'size : 932,308,473'. After running fasterq-dump without any other options you will have these fastq-files in your current directory: 'SRR341578_1.fastq' and 'SRR341578_2.fastq'. Each having a file-size of 2,109,473,264 bytes. In this case we have inflated the accession by a factor of approximately 4. But that is not all, the tool will need aproximately the same amout as scratch-space. As a rule of thumb you should have about 8x ... 10x the size of the accession available on your filesystem. How do you know how much space is available? Just run this command on linux or mac:
$df -h .
Under the 4th column ( 'Avail' ), you see the amount of space you have available.
Filesystem Size Used Avail Use% Mounted on
server:/vol/export/user 20G 15G 5.9G 71% /home/user
This user has only 5.9 Gigabyte available. In this case there is not enough space available in its home directory. Either try to delete files, or perform the conversion to a different location with more space.
If you want to use for instance a virtual 'RAM-drive' as scratch-space: (If you have such a device and how big it is, dependes on your system-admin!)
$df -h /dev/shm
If you have enough space there, run the tool:
$fasterq-dump SRR341578 -t /dev/shm
From version 2.11.4 on fasterq-dump checks if enough space is available for the output-file and the temporary files. This size-check is enabled by default, but can be explicitly turned off.
example: $fasterq-dump SRR000001 --size-check off
It is also possible to only perform the size-check, without the tool doing any conversion-work.
example: $fasterq-dump SRR000001 --size-check only
However the tool can not always detect the available space, especially if quotas are set. Because of that it is possible to manually specify the amount available.
example: $fasterq-dump SRR000001 --disk-limit 100GB
In case the temporary files and the output are on different filesystems, it is possible to specify the available disk-space separately for the temporary files and the output-file(s).
example: $fasterq-dump SRR000001 -t /dev/shm --disk-limit-tmp 8GB --disk-limit 100GB
In order to give you some information about the progress of the conversion there is a progress-bar that can be activated.
$fasterq-dump SRR341578 -t /dev/shm -p
The conversion happens in multiple steps, depending on the internal type of the accession. You will see either 2 or 3 progress bars after each other. The full output with progress-bars for a cSRA-accession like SRR341578 looks like this:
lookup :|-------------------------------------------------- 100.00%
merge : 13255208
join :|-------------------------------------------------- 100.00%
concat :|-------------------------------------------------- 100.00%
spots read : 7,549,706
reads read : 15,099,412
reads written : 15,099,412
for a flat table like SRR000001 it looks like this:
join :|-------------------------------------------------- 100.00%
concat :|-------------------------------------------------- 100.00%
spots read : 470,985
reads read : 470,985
reads written : 470,985
Because we have changed the defaults to be different and more meaningful than fastq-dump, here is a list of equivalent command-lines, but fasterq-dump will be faster.
fastq-dump SRRXXXXXX --split-3 --skip-technical
fasterq-dump SRRXXXXXX
fastq-dump SRRXXXXXX --split-spot --skip-technical
fasterq-dump SRRXXXXXX --split-spot
fastq-dump SRRXXXXXX --split-files --skip-technical
fasterq-dump SRRXXXXXX --split-files
fastq-dump SRRXXXXXX
fasterq-dump SRRXXXXXX --concatenate-reads --include-technical
Here are some important differences to fastq-dump:
- The
-Z|--stdoutoption does not work for split-3 and split-files. The tool will fail in these cases. - There is no
--gzip|--bizp2option, you have to compress your files explicitly after they have been written. - There is no
-Aoption for the accession; just specify the accession or the absolute path directly. -
fasterq-dumpdoes not take multiple accessions, just one. - There is no
-N|--minSpotIdand no-X|--maxSpotIdoption.fasterq-dumpprocesses always the whole accession, although it may support partial access in future versions.
To approach feature parity with fastq-dump, fasterq-dump supports a flexible defline. That means you can supply a user-defined defline for the the sequence and quality sections of FASTQ. There are 2 new commandline-parameters for that:
--seq-defline FORMAT and --qual-defline FORMAT
The format is a text that may contain these variables:
$ac ... the accession
$sn ... the spot-name
$sg ... the spot-group
$si ... the spot-id ( the number of the spot )
$ri ... the read-id ( the number of a read within a spot )
$rl ... the read-length
The accession, spot-id, read-id, and read-length are always available - but the spot-group and/or spot-name might be missing or empty. If a variable is missing or empty it does not produce an error - it will be omitted from the defline.
Please be aware that if the tool is used from within a shell-script, the format-string must be escaped to keep the shell from interpreting the variable names.
If no user-define is given to the tool, the following defaults are used:
FASTQ:
if not splitting @$ac.$si $sn length=$rl/+$ac.$si $sn length=$rl
if splitting: @$ac.$si/$ri $sn length=$rl/+$ac.$si/$ri $sn length=$rl
FASTA:
if not splitting >$ac.$si $sn length=$rl
if splitting: >$ac.$si/$ri $sn length=$rl
Be careful to choose the correct first character @/+/> based on the desired output (FASTQ/FASTA), as the tool will not correct it.
example for a shorter defline ( just containing the spot-name ) :
$sam-dump SRRXXXXXX --seq-defline '@$sn' --qual-defline '+$sn'
For every version newer and including 3.0.5 of the sra-toolkit, the fasterq-dump tool can handle PacBio accessions. There is no commandline switch neccessary to enable this feature. These PacBio accessions may or may not contain a consensus table in addition to the regular sequence table. Fasterq-dump will produce its output from the consensus table if it is present; otherwise, it will use the regular sequence table. If the user wants the output sourced from the sequence table, even if a consensus table is present, the following command can be used:
fasterq-dump SRRXXXXXX --table SEQUENCE
For every version newer and including 3.0.5 of the sra-toolkit, the fasterq-dump tool has some new functionality regarding references.
The new option '--fasta-ref-tbl' extracts references used to align the accession in FASTA-format. This option only works on accessions which are aligned, and because of that have a reference table. Each reference is extracted into one single FASTA-record in the output file. The output file is named after the accession with the extension 'ref.fasta' appended.
fasterq-dump SRRXXXXXX --fasta-ref-tbl This command produces a single file named 'SRRXXXXXX.ref.fasta'.
fasterq-dump SRRXXXXXX --fasta-ref-tbl -Z This command produces the same output on stdout.
fasterq-dump SRRXXXXXX --ref-report This command enumerates the references used by the accession on stdout. If the accession does not use references, the command will fail with an error message and return a non-zero error code. It can be used to test if an accession does contain references and is aligned.
fasterq-dump SRRXXXXXX --fasta-ref-tbl --internal-ref This command extracts only internal references into the output file. Internal references are non-standard scaffoldings the submitter included in the submission and the bases of them are stored in the accession.
fasterq-dump SRRXXXXXX --fasta-ref-tbl --external-ref This command extracts only external references into the output file. External references are canonical RefSeq accessions that are used in the accession. The bases of these references are not stored in the accession.
fasterq-dump SRRXXXXXX --fasta-ref-tbl --ref-name NC_011752.1 This command extracts only the named reference. The '--ref-name' option can be used multiple times per command. If a reference is not found, the option is ignored. If none of the requested references is found, an empty file is produced. Each reference used by an accession can be named in 2 ways: the canonical name like 'NC_001133.9' or the user-supplied name like 'I' or 'chr1'. The user supplied names are non-standard and specific to each accession. The user can use the '--ref-report' option to inspect the names used. Both names can be used to extract a specific reference. By default the canonical name is used in the defline of the FASTA-output. This default can be overwritten with the 'use-name' option. If this option is used, the submitter-supplied name is used in the defline of the FASTA-records.
There is one more option: '--fasta-concat-all'. This option is designed to be used on RefSeq accessions not on regular SRA accession. This option produces one single FASTA-record for the whole RefSeq accession.