Preprocessing methylation data
There are three types of methylation data.
- RRBS: Reduced-Representation Bisulfite Sequencing
- WGBS: Whole Genome Bisulfite Sequencing
- mCRF: methylCRF
cell_lines=("E003" "E004" "E005" "E006" "E007" "E011" "E016" "E038" "E047" "E066" "E087" "E114" "E116" "E117" "E118" "E119" "E120" "E123")
methylation_types=("WGBS" "WGBS" "WGBS" "WGBS" "WGBS" "WGBS" "WGBS" "mCRF" "mCRF" "WGBS" "RRBS" "RRBS" "RRBS" "RRBS" "RRBS" "RRBS" "RRBS" "RRBS")
for i in {0..17}
do
wget -b "https://egg2.wustl.edu/roadmap/data/byDataType/dnamethylation/${methylation_types[i]}/FractionalMethylation_bigwig/${cell_lines[i]}_${methylation_types[i]}_FractionalMethylation.bigwig"
done
The raw methylation data is in BigWig format. We can easily convert BigWig to BedGraph, which is a simpler format of Bed (only containing 4 columns), by code below.
wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bigWigToBedGraph
./bigWigToBedGraph E123_RRBS_FractionalMethylation.bigwig E123_RRBS_FractionalMethylation.bedGraph
Before using bedtools, we need to sort the BedGraph file.
sort -k1,1 -k2,2n E123_RRBS_FractionalMethylation.bedGraph > E123_RRBS_FractionalMethylation.bedGraph.sorted
Then, we are ready to map or intersect!
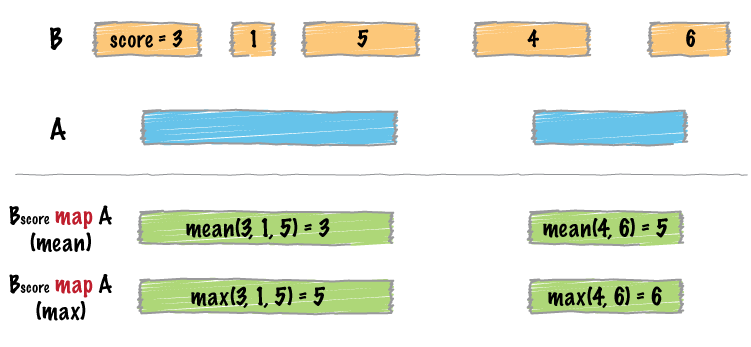
bedtools map -a tss_valid_chr_100bp.bed.sorted -b E123_RRBS_FractionalMethylation.bedGraph.sorted -c 4 -o mean
The option -c needs the column number of values and -o needs what method to be used. We will use 4 and mean, respectively.
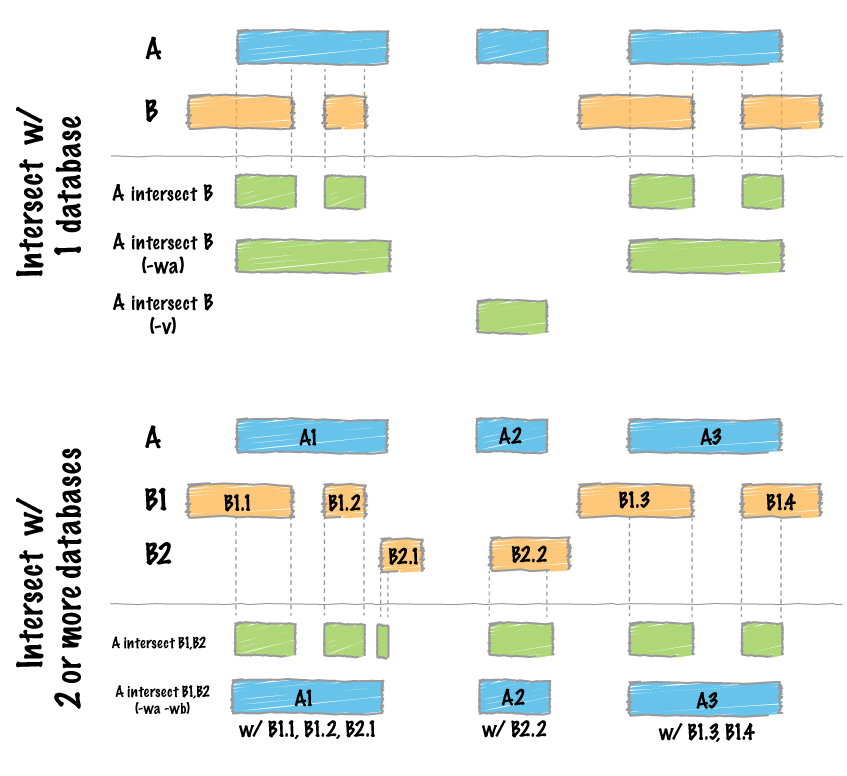
bedtools intersect -wa -wb -a E123_RRBS_FractionalMethylation.bedGraph.sorted -b simple_tss.sorted > E123_RRBS_FractionalMethylation.bedGraph.sorted.intersected
The command intersect is used for writing all CG positions regardless of bins. The option -wa and -wb are needed to write original entries of A and B files.