{kind=link}
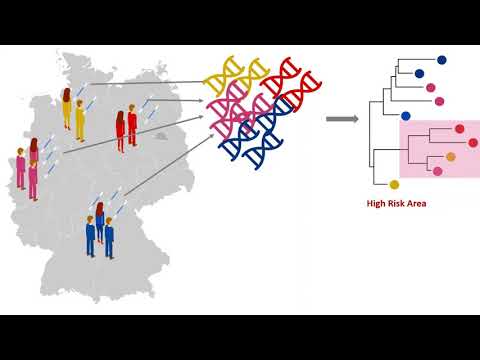
Our project CoroNet wants to identify high-risk areas where the virus is spreading very fast. We want to use the genetic information of the virus and the similarities between them to identify hotspots with fast evolving virus. Our goal is to slow down the infection spread of the virus and to flatten the curve when we are able to identify early the high-risk areas and isolate them from other regions. We use genomic sequences from GISAID to build a phylogenetic tree of the virus. From that, we can infer the direction of the spread by comparing the virus sequences taken from patients.
You need python and R installed. You will need the following python packages:
- matplotlib
- basemap
- geopy
- Biopython
and the following R packages
- ape
- ggsci
Additionally you will need the following commandline tools
MAFFT for constructing multiple sequence alignments (https://mafft.cbrc.jp/alignment/software/) RAxML-NG for building phylogenetic trees (https://academic.oup.com/bioinformatics/article/35/21/4453/5487384)
The notebook analyzes the Covid-19 spread through sequence evolution. The number of sequences in the maps does not reflect the number of cases because there iss only a very(!) small subset of patients for which viral sequences are obtained and those sequences are also publically available. For the connections between places, we use the genomic distances (number of basepair substitutions). This way, we can track the virus spread back in time.