
This is is the denovo pipeline from the Sequana projet
| Overview: | a de-novo assembly pipeline for short-read sequencing data |
|---|---|
| Input: | A set of FastQ files |
| Output: | Fasta, VCF, HTML report |
| Status: | production |
| Citation: | Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352 |
sequana_denovo is based on Python3, just install the package as follows:
pip install sequana --upgrade
You will need third-party software such as fastqc. Please see below for details.
The following command will scan all files ending in .fastq.gz found in the local directory, create a directory called denovo/ where a snakemake pipeline is stored. Depending on the number of files and their sizes, the process may be long:
::
sequana_denovo --help sequana_denovo --input-directory DATAPATH
This creates a directory with the pipeline and configuration file. You will then need to execute the pipeline:
cd denovo sh denovo.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s denovo.smk -c config.yaml --cores 4 --stats stats.txt
Or use sequanix interface.
This pipelines requires the following executable(s):
- spades
- busco
- bwa
- khmer : there is not executable called kmher but a set of executables (.e.g .normalize-by-median.py)
- freebayes
- picard
- prokka
- quast
- spades
- sambamba
- samtools
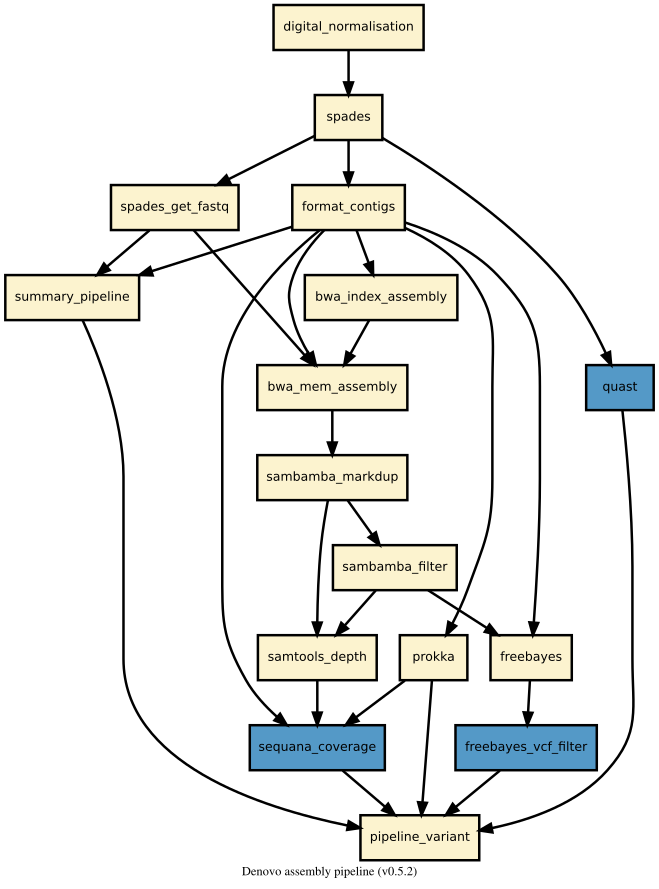
Snakemake de-novo assembly pipeline dedicates to small genome like bacteria. It is based on SPAdes. The assembler corrects reads and then assemble them using different size of kmer. If the correct option is set, SPAdes corrects mismatches and short INDELs in the contigs using BWA.
The sequencing depth can be normalised with khmer. Digital normalisation converts the existing high coverage regions into a Gaussian distributions centered around a lower sequencing depth. To put it another way, genome regions covered at 200x will be covered at 20x after normalisation. Thus, some reads from high coverage regions are discarded to reduce the quantity of data. Although the coverage is drastically reduce, the assembly will be as good or better than assembling the unnormalised data. Furthermore, SPAdes with normalised data is notably speeder and cost less memory than without digital normalisation. Above all, khmer does this in fixed, low memory and without any reference sequence needed.
The pipeline assess the assembly with several tools and approach. The first one is Quast, a tools for genome assemblies evaluation and comparison. It provides a HTML report with useful metrics like N50, number of mismatch and so on. Furthermore, it creates a viewer of contigs called Icarus.
The second approach is to characterise coverage with sequana coverage and to detect mismatchs and short INDELs with Freebayes.
The last approach but not the least is BUSCO, that provides quantitative measures for the assessment of genome assembly based on expectations of gene content from near-universal single-copy orthologs selected from OrthoDB.
| Version | Description |
|---|---|
| 0.11.1 |
|
| 0.11.0 |
|
| 0.10.0 |
|
| 0.9.0 |
|
| 0.8.5 |
|
| 0.8.4 |
|
| 0.8.3 |
|
| 0.8.2 |
|
| 0.8.1 | |
| 0.8.0 | First release. |