
Esse projeto foi feito como desafio final da Imersão de Dados da plataforma Alura. A propósta do projeto é análisar um conjunto de dados de pesquisa de medicamentos. O objetivo é passar por todas as etapas de um projeto de Machine Learning, desde a obtenção dos dados até a resolução do problema proposto. Foram usadas técnicas de Data Engineering, Exploratory Data Analysis e Machine Learning.
Como o prazo para entrega da solução foi curto, o princípal desafio foi entregar uma análise consideravelmente completa em pouco tempo. As análises e os resultados obtidos aqui podem ser considerados como um primeiro ciclo da metodologia CRISP DM - Cross Industry Standard Process for Data Mining*(Mais informações na bibliografia). Nesse método o projeto é visto como cíclico. Em cada ciclo são feitos incrementos e novas análises que possam aperfeiçoar a análise final.
Esse documento serve como um guia de estudo. A descrição das etapas estão inclusas nas próximas sessões.
Após cinco dias de estudos sobre as etapas de um projeto de Ciência de Dados, foi proposto um desafio para a análise de um conjunto de dados da área de Drug Discovery. O método de solução do problema ficou em aberto. Portanto, o nível de análise deveria ser escolhido pelo participante.
Foram disponibilizados dois conjuntos de dados. O primeiro conjunto agrupa diversas informações sobre determinados experimentos em culturas de células específicas. O segundo conjunto agrupa os resultados desses experimentos. O conjunto de resultados é dividido em 206 mecanismos de ação distintos e a sua classificação para ativação(1: ativa o mecanismo de ação, 0: não ativa o mecanismo de ação).
Pela alta granularidade desses mecanismos de ação, a análise desse projeto foi feita de maneira agregada. O problema escolhido foi o de desenvolver um modelo de Machine Learning que seja capaz de prever se determinada cultura de célular terá alguma ativação em pelo menos um dos mecanismos de ação.
A solução do problema foi feita através de um Modelo de Machine Learning. Esse modelo deve capaz de prever a classificação de uma cultura de célula em relação a ativação de pelo menos um mecanismo de ação. Além disso, o modelo final tem de prever a variável resposta de maneira mais precisa que um modelo aleatório.
-
Granularidade: Por cultura de célula;
-
Tipo do Problema: Classificação;
-
Método de Entrega: Modelo de Machine Learning.
Na etapa de Feature Engineering foram levantadas algumas hipóteses. Essas hipóteses foram análisadas durante a Análise Exploratória de Dados e geraram alguns Insights. Sistematizei a lógica por trás da ativação dos mecanismos de ação através de um mapa mental:
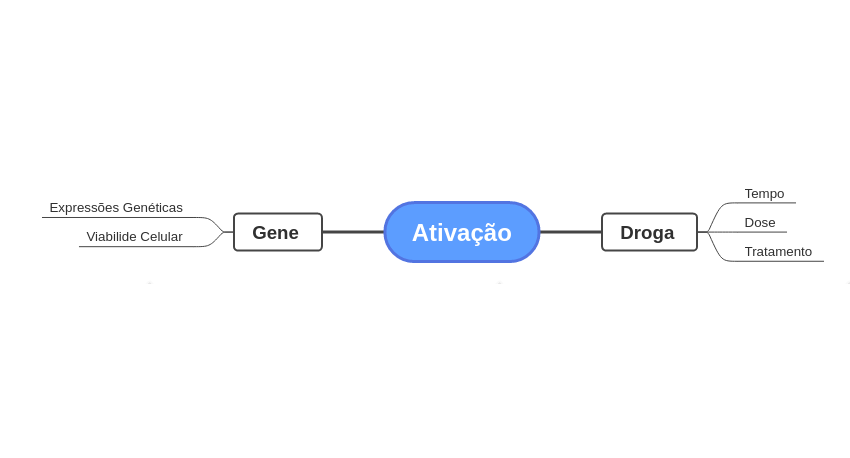
Essa visualização facilita o entendimento do problema. É possível entender como as variáveis são agrupadas em relação a variável resposta.
Como não sou um profissíonal da área, a análise desse resultado não é tão simples. A análise macro desse problema revelou que o período de efeito da droga tem pouca correlação com o número de ativações. Com as informações referentes ao tempo de efeito balanceadas, a quantidade de ativações seguiu o mesmo padrão para todos os tempos análisados.

Durante a etapa de entendimento, levantei a possibilidade da dose D2 ser superior a D1. Portanto a hipótese de que D2 ativaria mais que D1 foi colocada. No entanto, a análise demostrou que o tipo de dose tem pouca correlação com as ativações.

A análise das variáveis categóricas demostrou baixa correlação. A única variável que se correlacionou de maneira minimamente satisfatória com as ativações foi a Tratamento. É de se esperar que o Tratamento e as Ativações tenham maior correlação. Isso pode ser afirmado pois apenas tratamentos com droga levam a ativação.

Outro aspecto interessante é a correlação entre os valores de Viabilidade Celular e Expressão Genética. É possível perceber que existe um padrão na correlação entre essas duas variáveis. A correlação da expressão genética com as viabilidades celulares é sempre a mesma para os valores de c-#. Por exemplo, Se a correlação entre um g-# e um c-# é alta, todos os demais valores de c-# para esse g-# repetirão esse padrão alto. O motivo disso pode ser explicado por algum especialista da área ou através de uma análise específica desse efeito.

Para a aplicação do modelo, algumas etapas foram cumpridas:
- Divisão dos dados entre Treino e Teste - Os dados de treino são usados para criar o modelo. Os dados de teste servem para entender o quão bem o modelo generaliza;
- Encoding das variáveis categóricas - Transformação das variáveis categóricas em variáveis numéricas.
- Feature Selection - A explicação mais simples é sempre preferível do que a mais complexa. Esse é o princípio da navalha de Occam. Soluções complexas são mais propensas a erros. A simplificação do problema deve ser o objetivo sempre. Para os dados em questão foi utilizado um modelo de Random Forest para classificar a importância das features em relação ao projeto. Apenas as 25% features mais bem classificadas foram utilizadas.
Cumprida essas etapas, a próxima etapa é modelar o problema. O modo de solução escolhido foi um modelo de Classificação. Esse modelo deve prever dados nunca vistos de maneira satisfatória. Para isso, foram feitos testes de seis algorítmos de Classificação específicos:
- 1. Regressão Logística
- 2. KNN
- 3. GaussianNB
- 4. LGBMClassifier
- 5. RandomForestClassifier
- 6. CatBoostClassifier
Foi utilizado o método de Validação Cruzada para o cálculo das métricas. A Validação Cruzada separa os dados em folds. Cada fold desempenha o papel de conjunto de teste em cada ciclo, e os demais folds são usados para a modelagem. Esse procedimento permite reduzir o viés e aumentar a capacidade de generalização do modelo. Os desempenhos foram os seguintes:

Os principais modelos tiveram desempenho semelhante. Apesar de ter um desempenho superior em relação aos demais, o CatBoostClassifier demanda muito processamento e tempo. Para fins desse projeto, o modelo escolhido foi o LGBMClassifier.
LGBMClassifier escolhido, os parâmetros de entrada do modelo foram tunados para buscar um maior desempenho. Com o modelo final pronto, é hora de testar o seu poder de generalização com os dados de teste, seguindo o método de Validação Cruzada:

- Acurácia: A taxa de acerto, independente da classe em questão;
- Precisão: Daqueles o modelo previu como positivo, quantos são realmente positivos?;
- Recall: Daqueles que são positivos, quanto o modelo previu?;
- Specificity: Quão bom o modelo é bom em prever a classe negativa?
Os resultados do modelo pouco informam sem uma base de comparação. Para isso, foram criados duas baselines: A primeira com valores aleatórios entre 0 e 1, a segunda que determina que todos os valores são 1. Os resultados foram os seguintes:

Algumas conclusões podem ser tiradas desses dados:
-
O modelo foi mais acurado que ambas as baselines. A taxa de acerto do modelo foi de 69.13%;
-
O modelo foi mais preciso que ambas as baselines. 66.39% dos dados que o modelo previu como ativados, realmente foram ativados;
-
O modelo teve o Recall praticamente identico ao modelo que "chutou" todos os valores como ativados. O recall é a taxa que demonstra a porcentagem de dados que realmente são positivos e o modelo previu. Como a baseline chutou todos os valores como ativados, ele acertou em todas as vezes. Apesar disso, o modelo usado chegou bem próximo desse valor, com um Recall de 99.49%;
-
A Specificity do nosso modelo foi superior a baseline_todos1. Com uma de taxa de 22.30%, apesar do modelo acertar em aproximadamente 100% daqueles que são positivos, ele ainda tem uma taxa de acerto dos valores negativos. No entanto a taxa ainda é muito baixa. Os próximos ciclos devem ser focados em aumentá-la.
O modelo de LGBMClassifier permitiu um aumento no poder de previsão em relação a ativação das culturas celulares. O modelo se mostrou superior às baselines aleatória e a todos_1. Isso pode ser interpretado como um avanço. No entanto, novas etapas devem ser enfrentadas para melhorar ainda mais esse resultado.
-
Esse projeto foi desafiador para mim. Comecei a produzi-lo no dia anterior a sua publicação e estou bastante feliz com o resultado obtido. O curto prazo para entrega e a grande quantidade de dados me ensinou que é possível entregar uma solução, ainda que básica, de um problema;
-
Durante as pesquisas, consegui me aprofundar em conceitos biológicos e de Drug Discovery que não conhecia. Irei me aprofundar nesse tema para trabalhar de maneira mais avançada nesse mesmo conjunto de dados;
-
Consegui aperfeiçoar as ferramentas técnicas de manipulação e visualização de dados. Também aprendi ferramentas novas, como o Crosstab e os métodos de balanceamento de dados(apesar de não ter utilizado no projeto).
Os resultados foram melhores do que previsões aleatórias e "chutes". No entanto, as métricas ainda estão abaixo do que podem ser. Os próximos passos para esse projeto são:
-
Trabalhar com novos ciclos do CRISP;
-
Me aprofundar nos conceitos Biológicos;
-
Fazer uma Análise Exploratória de Dados mais avançada para a criação de Insights;
-
Testar novos modelos de Machine Learning.
Drug discovery: passado, presente e futuro.
Expressão gênica: o caminho da informação biológica.