![]()

A module to estimate contamination level from diploid variant calls. This is heavily inspired by Dcon.
We are hypothesizing the contamination level would be between
Pseudo code:
n = total_count
x = alt_allele_count
contam = 0
max_log_likelihood = -inf
for contam_level in all_contamination_level:
p = expected_alt_fraction_for_the_given_contamination_level
log_likelihood = sum(binom_loglik(n, x, p) for all_variants)
if log_likelihood > max_log_likelihood:
contam = contam_level
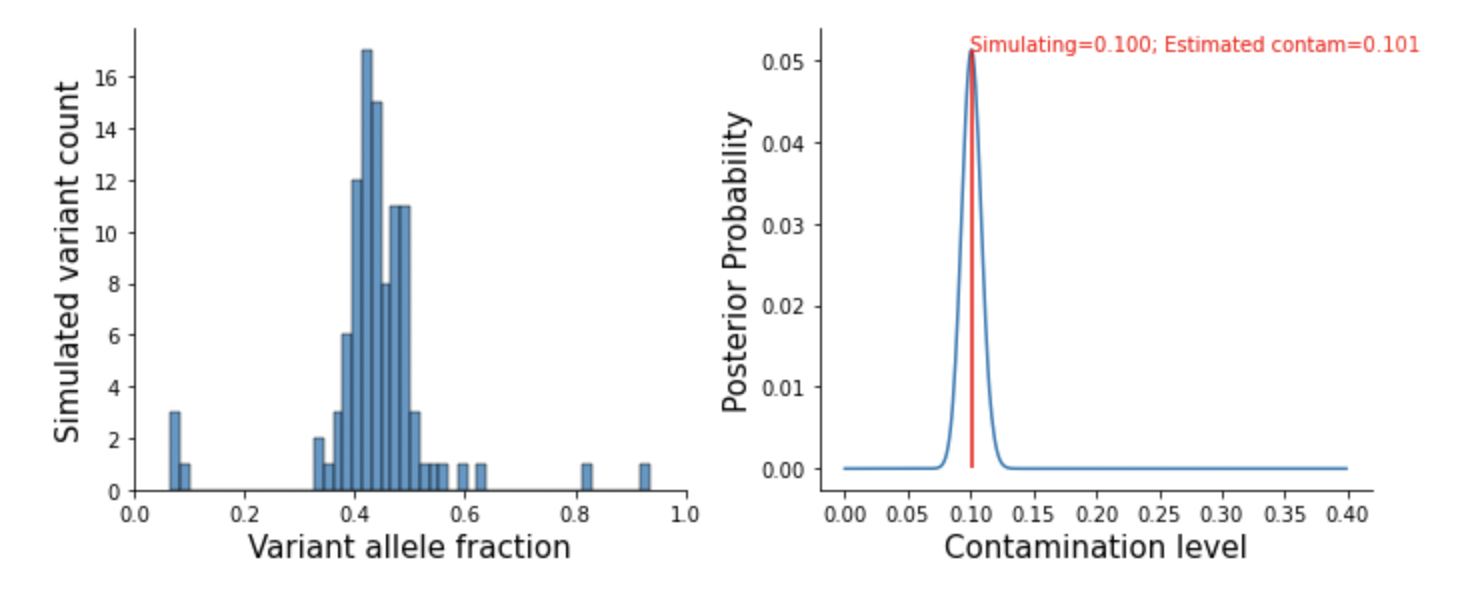
A simulated study at here.
For a homozygous variant, the probability of observing the expected variant-allele-count (
where

For a heterozygous variant, the probablity of observing the expected variant-allele-count (
-
$(1 - c)/2$ , when a low alternate allele frequency is observed because of the contamination -
$(1 - c)$ , when a homozygous variant being called as a heterozygous variant because of the contamination -
$(0.5 + c)$ , when the contamination looks like the alternate allele, such that the alternate allele frequency is higher than expected -
$(0.5 - c)$ , when the contamination looks like the reference allele, such that the alternate allele frequency is lower than expected -
$c$ , when the contamination itself is called as low variant frequency heterozygous variant
After evaluating these cases, we will pick the highest probability event when summing the log likelihoods for the given contamination level.
We also wrote the code in rust.
To run the code:
$ cargo run -- -i data/test.vcf -d debug_json
or:
$ cargo install --path .
$ target/release/diploid-contam-estimator
Douglas Wu <wckdouglas@gmail.com>
Estimating contamination level from a diploid VCF file
The program assume we are dealing with a diploid genome, and using the
deviation of allelic balance from the expected allelic frequence for homozygous
or heterozygous variant calls to compute a contamination value.
For homozygous variants, we deviation from allelic frequency of 1 is all introduced by
contaminaion.
For heterozygous variants, it is a little more complex, because it could be due to:
1. contamination that doesn't look like the HET ALT allele: we expect lower HET alt allele
frequency
2. contamination that doesn't look like the HOM ALT allele: we expect High HET alt allele
frequency
3. contamination that looks like the ALT allele: we expect higher alt allele frequency
4. contamination that looks like the REF allele: we expect lower alt allele frequency
5. contamination being called as ALT
USAGE:
diploid-contam-estimator [OPTIONS] --in-vcf <in_vcf>
OPTIONS:
-d, --debug-json <debug_json>
A json output file for storing all intermediate log prob
-h, --help
Print help information
-i, --in-vcf <in_vcf>
A diploid vcf file for estimating contamination
-m, --min-depth <depth_threshold>
Minimum depth for a variant to be considered (i.e. DP tag) [default: 0]
-o, --out-json <out_json>
A json output file for storing the maximum likelihood contam level for the vcf file
--snv-only
Only use SNV (ignore indel) for contamination estimations
-v, --debug-variant-json <debug_variant_json>
A json output file for storing all input variants used for calculation
-V, --version
Print version information