Phylogenomics from Low-coverage Whole-genome Sequencing We present a novel WGS-based pipeline for extracting essential phylogenomic markers through rapid genome assembling from low-coverage genome data, employing a series of computationally efficient bioinformatic tools. We tested the pipeline on a Hexapoda dataset and a more focused Phthiraptera dataset (genome sizes 0.1‒2 Gbp), and further investigated the effects of sequencing depth on target assembly success rate based on raw data of six insect genomes (0.1‒1 Gbp). Each genome assembly was completed in 2‒24 hours on desktop PCs. We extracted 872‒1,615 near-universal single-copy orthologs (BUSCOs) per species. This method also enables development of ultraconserved element (UCE) probe sets; we generated probes for Phthiraptera based on our WGS assemblies, containing 55,030 baits targeting 2,832 loci, from which we extracted 2,125‒2,272 UCEs. We also showed that only 10‒20× sequencing coverage was sufficient to produce hundreds to thousands of targeted loci from BUSCO sets, and even lower coverage (5×) was required for UCEs. Our study demonstrates the feasibility of conducting phylogenomics from low-coverage WGS for a wide range of organisms. This new approach has major advantages in data collection, particularly in reducing sequencing cost and computing consumption, while expanding loci choices.
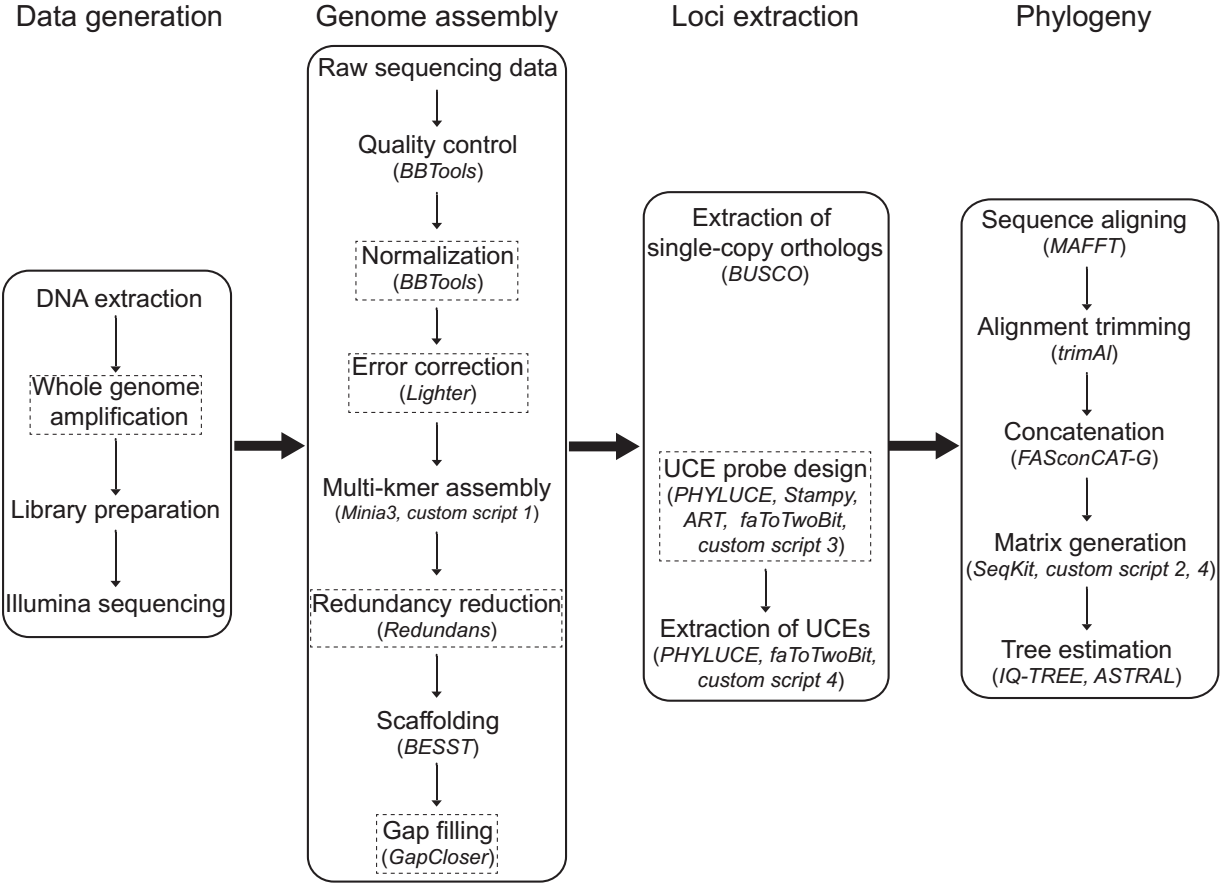
All four bash scripts are only tested in Centos operating system, and may be available for other Linux systems. Their functions are described below.
- script1_Genome_assembly.sh: Script 1 performs main rapid genome assembly steps, i.e. read quality trimming and normalization (BBtools), multi-k-mer assembly (Minia3), reduction of heterozygous contigs (Redundans), scaffolding (BESST), gap filling (GapCloser) and BUSCO assessment (BUSCO).
- script2_BUSCO_extraction.sh: Script 2 extracts single-copy orthologs (BUSCOs) from previous BUSCO assessments and generates nuclotide/protein alignment matrices of 50%-100% completeness (MAFFT, trimAl and FASconCAT-G) and partitioning schemes for phylogenetic analyses.
- script3_UCE_probe_design.sh: Script 3 can design UCE probe with tools art, BBtools, faToTwoBit and stampy.
- script4_UCE_extraction.sh: Script 4 extracts UCE loci from genome assemblies and generates alignment matrices of 50%-100% completeness and partitioning schemes for phylogenetic analyses. Tools faToTwoBit and FASconCAT are required.
Requirements
Some bioinformatic tools are neccessary for above scripts. Most of them are recommended to be added into the environmental paths. Softwares, versions and source ate listed below.
BBTools v38.32 (https://sourceforge.net/projects/bbmap/)
Minia v3.2.4 (https://github.com/GATB/minia)
Redundans v0.14c (https://github.com/lpryszcz/redundans)
Minimap2 v2.12 (https://github.com/lh3/minimap2)
Samtools v1.9 (http://www.htslib.org/)
BESST v2.2.8 (https://github.com/ksahlin/BESST)
GapCloser v1.12 (http://soap.genomics.org.cn/)
BUSCO v3.0.2 (http://busco.ezlab.org/)
MAFFT v7.407 (https://mafft.cbrc.jp/alignment/software/)
trimAl v1.4.1 (http://trimal.cgenomics.org/)
FASconCAT-G v1.04 (https://github.com/PatrickKueck/FASconCAT-G)
PHYLUCE v1.6.6 (http://phyluce.readthedocs.io/en/latest/index.html)
faToTwoBit (http://hgdownload.soe.ucsc.edu/admin/exe/)
ART-20160605 (https://www.niehs.nih.gov/research/resources/software/biostatistics/art/index.cfm)
Stampy v1.0.32 (http://www.well.ox.ac.uk/project-stampy)
TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder)
To faciliate the use of our pipeline and simplify the installation of various packages, we also prepared the virtual mirror (.vmdk), including all the bioinformatic tools and custom scripts ready in the CENTOS 7.3 system. It can be directly opened by VMware or VirtualBox, which are often installed on Windows systems. Please download it here: https://1drv.ms/f/s!Ak7sQGBGqlguq1gdk1KKIR7uwAfj. More than 100 Gb (at least 50) disk space is recommented for its use. Below is related information on CENTOS system:
operating system: centos 7.3
passwords of accounts, i.e. root and zf: 1
Most packages are installed under /usr/local/bin, /home/zf/install/ etc. Environmental variables and paths can be checked by "vi ~/.bashrc" and "printenv". Most executables can be directly performed without typing the installation paths. UCE-related analyses should be executed by initiating conda environments (/home/zf/install/miniconda/envs/), such as phyluce, just type 'source activate phyluce'.
User manual
The requirements for each script have been described in the beginning of the bash script text. Values of variables/parameters, such as read length, tool path, number of threads etc., can be modified at the beginning of analyses following the script guidance.
Below are some notes for each script although more details have been described in the script.
Script1:
- Type 'sh script1_Genome_assembly.sh forward_reads_file reverse_reads_file', e.g. sh script1_Genome_assembly.sh 1.clean.fq.gz 2.clean.fq.gz. Adaptors must to be removed prior to the analyses.
- Most executables are recommended to be added into the environmental paths. Tools pigz, BBTools, Minia, redundans, Minimap2, samtools, BESST, GapCloser and BUSCO may be used and will be automatically checked prior to formal analyses in this script.
- The default starting kmer value is 21 and thus kmer values are 21, 21+20, 21+2*20....
- The statistics of assemblies generated in the asembly procedure are summerized in assembly.statistics
Script2:
- All the BUSCO results (run_* folders) are deposited in the same folder, e.g. BUSCOs/. Type 'sh script2_BUSCO_extraction.sh BUSCO_folder', e.g. sh script2_BUSCO_extraction.sh BUSCOs
- Tools TransDecoder, MAFFT, trimAl and FASconCAT-G are used in this script and will be automatically checked prior to formal analyses
- The final matrices, partition files and summary are placed in 5-loci_concat/
Script3:
- Type 'sh script3_UCE_probe_design.sh assemblies_folder', e.g. sh script3_UCE_probe_design.sh assemblies
- Copy all the genome assemblies to a folder (e.g. assemblies/) and replace assembly name using "SPECIES_NAME.fa"
- Tools art, BBtools, faToTwoBit and stampy, as well as PHYLUCE conda enviorment, are used and will be automatically checked prior to formal analyses in this script
Script4:
- Type 'sh script4_UCE_extraction.sh assemblies_folder probe_sequence', e.g. sh script4_UCE_extraction.sh assemblies probe.fasta
- Modify all genome assemblies endding with .fa ("SPECIES_NAME.fa") and copy them to the same folder
- Tools faToTwoBit and FASconCAT, as well as PHYLUCE conda enviorment, are used and will be automatically checked prior to formal analyses in this script. MAFFT and trimal are not necessary to install because they have been included in the PHYLUCE conda enviorment.
Citation
Feng Zhang, Yinhuan Ding, Chaodong Zhu, Xin Zhou, Michael C. Orr, Stefan Scheu, Yun-Xia Luan. 2019. Phylogenomics from Low-coverage Whole-genome Sequencing. Methods in Ecology and Evolution. DOI: 10.1111/2041-210X.13145
Contact
Please send emails to Dr. Feng Zhang (xtmtd.zf at gmail.com).