- Installation package
- Github: WinSNPGT.exe
- Figshare: WinSNPGT.exe
- CSDN: WinSNPGT.exe
- repository
- Github: Min-Zer0/WinSNPGT
- Gitee: Min-Zer0/WinSNPGT
The rapid development of sequencing technology and dramatic drop in the cost have led to the generation of massive amounts of data. However, most of the raw data are analyzed on linux systems, and the process of generating variant loci information from sequencing data is a challenge for researchers unfamiliar with linux systems.
We have developed a toolkit to call variant loci on the Windows system, WinSNPGT. It can obtain the genotypes of the raw sequencing data for the snp loci specified in our datasets.
The installation and use of this toolkit is described below.
We have developed a phenotype prediction platform, CropGS-Hub, which contains multiple high-quality datasets from important crops such as rice, maize and so on. These datasets were used as training sets to build models for phenotype prediction. Users can upload genotypes of their own samples to the platform for online phenotype prediction.
The WinSNPGT toolkit was developed to ensure that the genotypes uploaded by users match those in the training set for modeling so that bias in the prediction results can be avoided. Users can run this program on the Windows system to realize the whole process from sequencing files to getting genotypes by simple operation, which is very friendly for people who have little experience in linux operation.
-
java8 download link:Download Link1 | Download Link2 | Download Link3 | Download Link4
- Default installation
-
WinSNPGT download link:👉 Latest release package
- Double-click the exe installation package, select the installation path (
Chinese characters are not allowed) - After decompression is completed, the following prompt appears, indicating that the installation is successful:
- Double-click to enter the installation path WinSNPGT/
WinSNPGT:Startup icon (a shortcut will also generate on the desktop)
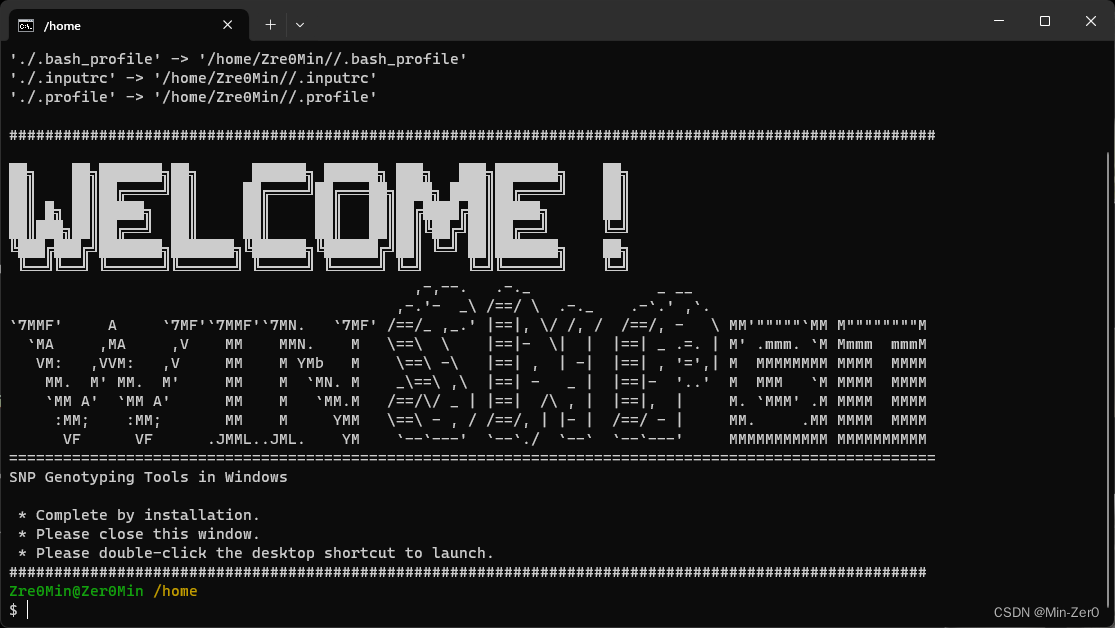
Input_Fastq/:Paired-end sequencing files should be moved as input into this folder
Result/:Folder where the results are output after the program is finished running
Reference_Genome:Folder where the reference genome file are placed (generally not used) - Double-click the exe installation package, select the installation path (


The example-data files are not included in the release package, you can download:example-data.tar.gz
The species of the example-data files is Oryza sativa, you can select the rice-related dataset (e.g: GSTP007 ~ GSTP009) in the toolkit to complete the genotyping.
- Move your raw sequencing data
(*.fastq.gz)or(*.fastq)to the path: ./Input_Fastq
- Double-click to run the program and a web pop-up will appear in the default browser
- There will also be a window running as a background program
Do not close the background program windowIt should be closed after the program is finished running or when restarting the program, you need to close this window
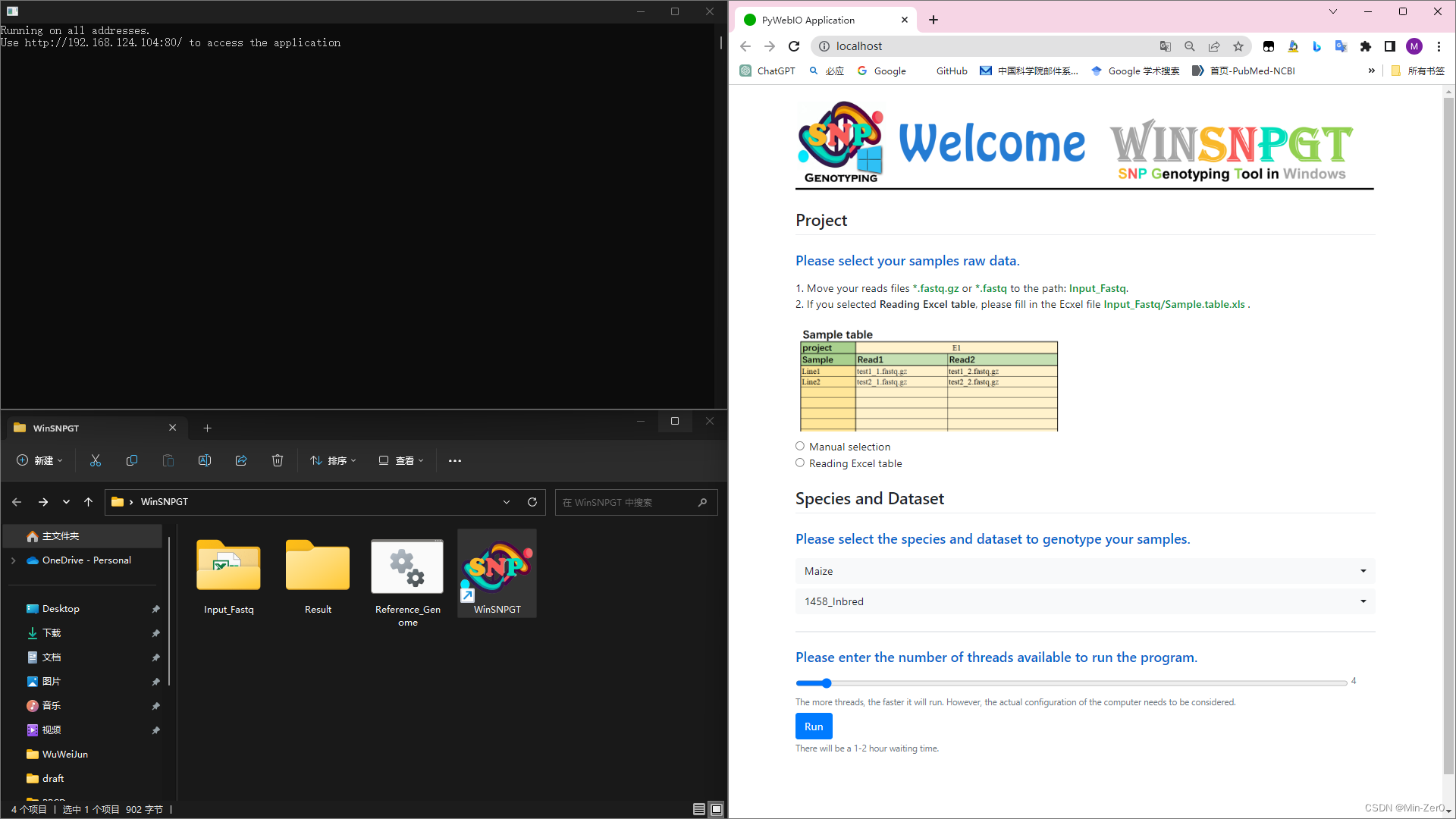
There are two ways to read raw reads files.
- Manual selection:
When there are few samples, it is recommended to quickly add and generate the form.- Enter project name which will be output file prefix
- Select the corresponding reads files and enter the sample name
- Click Add button to update the form if there are another samples to be genotyped
- Click Submit button after adding all samples to be genotyped and confirm the form is correct
Project name and sample names are limited to numbers and upper- and lower-case letters (replace spaces with underscores); sample names may not be all numbers.

- reading excel table
It is recommended to avoid errors in manual selection when there are too many samples.- Fill in the file Sample.table.xls under the path ./Input_Fastq in advance
Project name and sample names are limited to numbers and upper- and lower-case letters (replace spaces with underscores); sample names may not be all numbers.

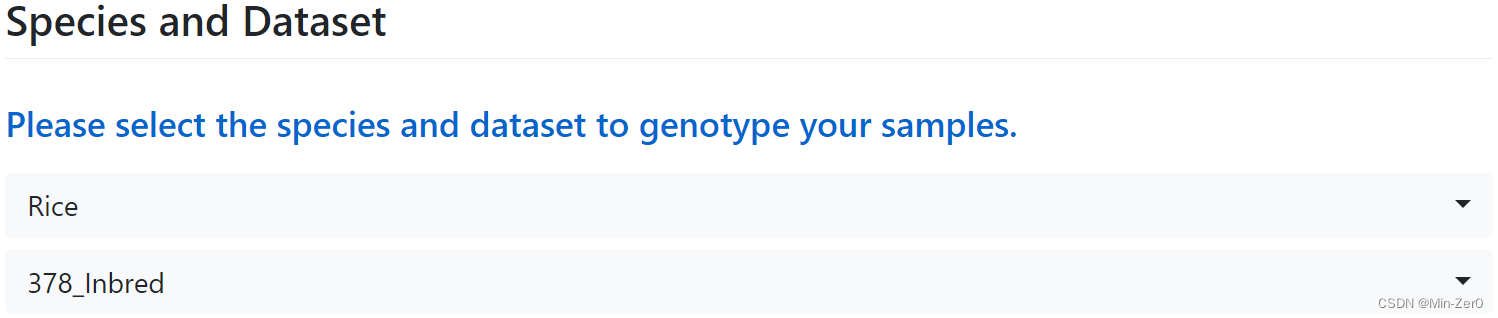
- Select the species of your samples to be genotyped
- Select the dataset corresponding to the model to be fitted
- Datasets available:CropGS-Hub Dataset
- The default number of threads is 4


- During normal operation, the program will automatically download the reference genome before entering the genotyping analysis process.
- Users can download it offline if they do not have an Internet connection.
- Download link:
- Maize (Zea mays):
- GSTP001_8652_Hybrid : Maize_8652_Hybrid
- GSTP002_5820_Hybrid : Maize_5820_Hybrid
- GSTP003_1458_Inbred : Maize_1458_Inbred
- GSTP004_1404_Inbred : Maize_1404_Inbred
- GSTP005_350_Inbred : Maize_350_Inbred
- GSTP006_1604_Inbred : Maize_1604_Inbred
- Rice (Oryza sativa):
- GSTP007_1495_Hybrid : Rice_1495_Hybrid
- GSTP008_705_Inbred : Rice_705_Inbred
- GSTP009_378_Inbred : Rice_378_Inbred
- Cotton (Gossypium hirsutum):
- GSTP010_1245_Inbred : Cotton_1245_Inbred
- Millet (Setaria italica):
- GSTP011_827_Inbred : Millet_827_Inbred
- Chickpea (Cicer arietinum):
- GTP012_2921_Inbred : Chickpea_2921_Inbred
- Rapeseed (Brassica napus):
- GSTP013_991_Inbred : Rapeseed_991_Inbred
- Soybean (Glycine max):
- GSTP014_2795_Inbred : Soybean_2795_Inbred
- Maize (Zea mays):
- After downloading the corresponding species and datasets, double-click Reference_Genome.bat, open the path to the file, and drag the offline downloaded file into it.
- This function can also be used to migrate species and datasets between different users without repeated downloads.
-
After the program is completed, the result file will be output in the Result/ directory.
- Standard format VCF (variant call format) file
- *.Genotype.txt (Sample genotyping matrix, the format is as follows)
CHROM POS Line 1 line 2 ... line N Chr1 128960 A . ... C Chr1 133137 C C ... T ... ... ... ... ... ... Chr12 321216 A A ... A Chr12 364257 A C ... C Chr12 364755 . . ... . ... ... ... ... ... ... -
Upload *.Genotype.txt to CropGS-Hub to complete subsequent analysis
- Phenotype Prediction PhenotypePrediction
- Crossing Design CrossingDesign
If there are some errors reported during the running of the program, please refer to the following scenarios to solve the problem:
- The
background program is not allowed to be closeduntil you get your results. - If you fill in the wrong information on the web page, you can
refresh and refill the interfacebefore running, and there is no need to restart the program or repeat moving files steps. Chinese characters are not allowedto appear inpath where winSNPGT is installed. Otherwise, the following error may occur:- If you chose the way to read excel table, you must
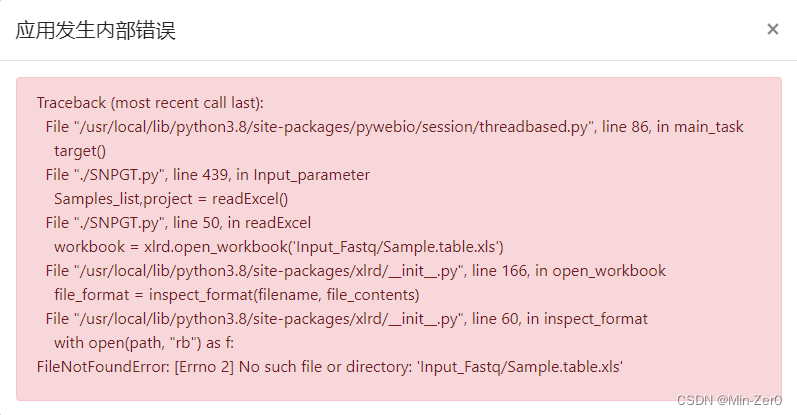
save and close the tableafter filling it, which means it cannot be kept open. Otherwise, the following error may occur:

The above are some possible causes of errors, if there are any other problems, welcome to contact us.
- Jie Qiu (qiujie@shnu.edu.cn)
- Min Zhu (zer0min@outlook.com)
- Jiaxin Chen (jxchen1217@gmail.com)