
![]()


Proteins from evolutionarily distant ancestors can have amino acid sequences which are so different that traditional sequence alignment methods can fail to detect any similarity. However similarity in the 3D structure of proteins often persists long after their sequences have diverged. It is possible to detect evolutionary similarity between proteins from distantly related organisms by examining their 3D structure.
MINRMS is a program for aligning two proteins by considering their 3D structure. (MINRMS also works with other kinds of biopolymers.) It reads two PDB files and tries to find a subset of residues from either molecule with similar shape. (PDBx/mmCIF and PDBXML files are not yet supported.) Earlier structural alignment programs used ad-hoc scoring functions to distinguish between possible solutions. However MINRMS finds alignments which minimize the root-mean-squared-distance (RMSD) between matched residues from either molecule. (See below.) Arguably, RMSD is the simplest structure alignment metric and is often quoted as a measure of alignment quality. Unlike earlier probabilistic programs, MINRMS performs an exhaustive search.
Generated alignments are stored in MSF format. (MSF files can be converted to and from other more popular alignment file formats (such as FASTA) using aligncopy.)
MINRMS documentation may be found online, and also here.
Documentation for other programs bundled with minrms can be found here..
minrms -fm 4 -of 8.0,0.33 \
-minN 0.33 -max-rmsd 3.5 -ir -r \
hemoglobin.pdb,"*.A" myoglobin.pdb
This will find the lowest-possible RMSD alignments between chain A of "hemoglobin.pdb" and all of "myoglobin.pdb", with the following constraints:
- Alignments which fail to match at least 33% of the amino acids in both protein will be ignored.
- Alignments which have an RMSD exceeding 3.5 Angstroms will be ignored.
- Alignments which (after optimal rotation) contain any pairs of residues separated by more than 8.0 Angstroms will be ignored. (This prevents the creation of alignments between distant portions of the molecules.)
- Alignments which fail to match any secondary structure (helices with helices or sheets with sheets) will be ignored. (If your PDB files lack HELIX or SHEET records, include the "-HS" argument.)
You can relax these constraints by ignoring the optional arguments above:
minrms -fm 4 -ir -r hemoglobin.pdb,"*.A" myoglobin.pdb
(This will run much more slowly and is not recommended.)
If either of the PDB files lack HELIX and SHEET secondary structure information, you must warn minrms by including the "-HS" argument.
The main minrms program is written in C++. A C++11 compliant compiler is required to build the executable (such ass GCC 4.9+ or CLANG 6+ or later).
(The bin/ subdirectory also includes some additional programs which are documented here. These programs are probably not very relevant for most users and can be ignored.)
Compilation and installation instructions can be found here.
If you find this program useful, please cite:
"MINRMS: an efficient algorithm for determining protein structure similarity using root-mean-squared-distance", Bioinformatics, 2003, 19(5):625-34, Jewett AI, Huang CC, Ferrin TE https://doi.org/10.1093/bioinformatics/btg035
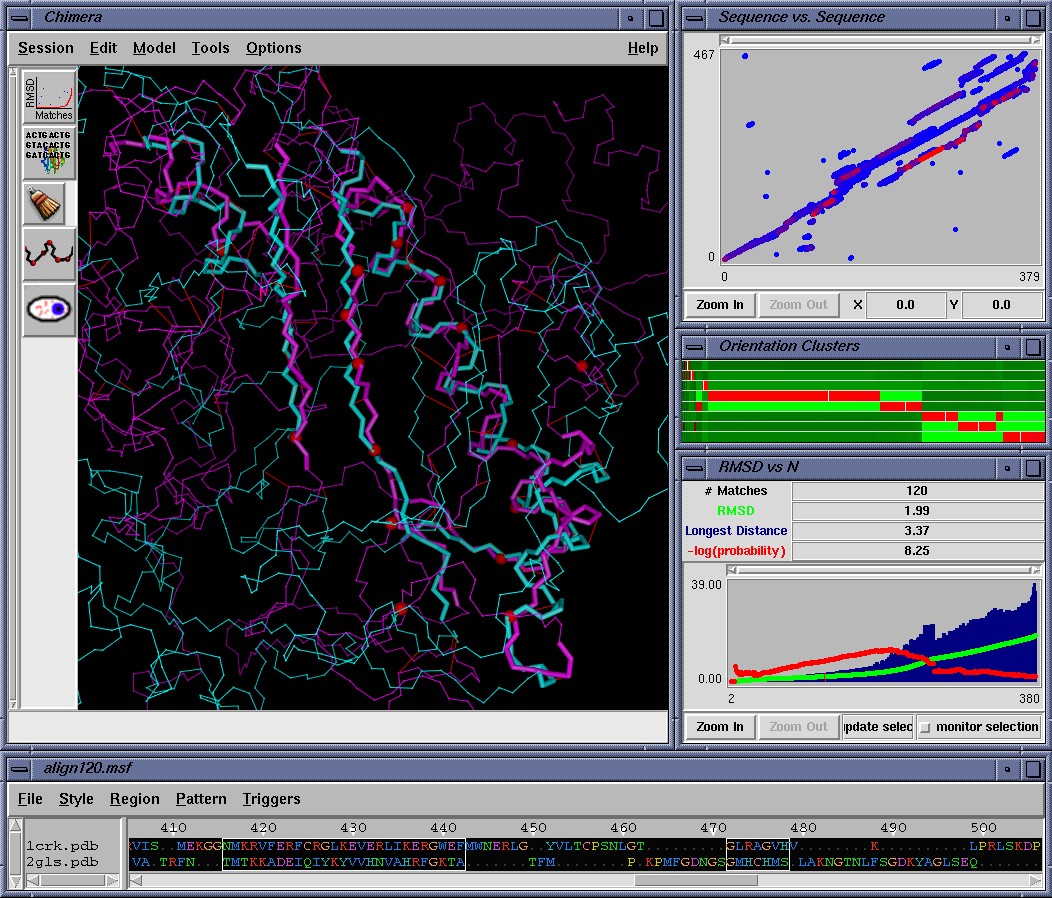
RMSD is a simple metric which is easy to interpret. However it does not indicate how many amino acids the two proteins have in common. In the absence of other criteria, choosing alignments on the basis of their RMSD favors alignments with very few residues in common, even if the two proteins have nearly identical shape. (This is because it's always possible to find an alignment with a lower RMSD by discarding the most distant pair of residues in an existing alignment.) As a result of this ambiguity, MINRMS will generate alignments of every possible size, each of which is optimal in RMSD (for alignments of that size). It is the responsibility of the user to choose between these alignments.
To do that, users can visually browse all of these alignments using the MinrmsPlot/AlignPlot menu option in Chimera., which is shown above. This will display each alignment and report it's Levitt and Gerstein's P_str score (which is plotted in red). The P_str scores can be used as a basis for selecting the optimal alignment, in addition to visual inspection. Alternatively the P_str scoring metric can also be calculated independently using the stand-alone msf2stat3d program which is included with this repository. (However we strongly recommend viewing the alignments in Chimera to make sure the alignment you selected is reasonable.)
In typical usage (when the recommended settings are used), MINRMS has a O(m^2 n^2) running time, where m and n are the number of amino acids in each protein. When no constraints are used, MINRMS has a running time of O(m^3 n^2).