Tutorial
This tutorial will highlight the key FuncDiv functions. Make sure you install the R package, if you have not already.
This tutorial will focus on several example files that are included in the FuncDiv GitHub repo.
First, clone the repository (if you did not already download it during installation).
Then open a new R session and navigate to the example data folder:
setwd("Path/to/FuncDiv/example_files/")These files are present in the example_files folder:
-
func_input.tsv.gz- Table of gene (KEGG ortholog) copy numbers per taxon (metagenome-assembled genomes) -
taxa_input.tsv.gz- Table of taxon abundances per sample -
contrib_input.tsv.gz- Table of combined gene copy numbers per taxon and taxon abundance per sample. This is in contributional format (an extremely long file), which is an alternative to inputting the two above tables. -
taxa.tree- Phylogenetic tree of all metagenome-assembled genomes
Read in all these files.
func_tab <- read.table("func_input.tsv.gz",
header = TRUE, sep = "\t", row.names = 1, check.names = FALSE)
abun_tab <- read.table("taxa_input.tsv.gz",
header = TRUE, sep = "\t", row.names = 1, check.names = FALSE)
contrib_tab <- read.table("contrib_input.tsv.gz",
header = TRUE, sep = "\t", stringsAsFactors = FALSE, check.names = FALSE)
in_tree <- ape::read.tree("taxa.tree")It can be helpful to take a look at example input objects if you are unsure how your own files should be formatted.
For instance, you can take a look at the first five rows/columns of the func_tab table like this:
func_tab[1:5, 1:5]
# SRR2912782_bin.17 14207_6_11 ERR688549_bin.7 20298_3_1 ERR1305881_bin.83
# K04749 0 0 0 0 0
# K16052 0 0 0 0 0
# K01956 1 1 1 1 1
# K17218 0 0 0 0 0
# K01474 0 0 0 0 0And the first few lines of contrib_tab:
head(contrib_tab)
# samp func tax tax_func_copy_number tax_abun
# SRR3546780 K04749 GCF_000010185 1 14.89
# SRR3498907 K04749 21673_4_91 1 1.97
# SRR3498907 K04749 SRR2155554_bin.21 1 6.98
# SRR3498907 K04749 SRR5056999_bin.11 1 9.06
# SRR3546779 K04749 GCF_000010185 1 23.74
# SRR3546776 K04749 GCF_000010185 1 16.97You can see the default alpha diversity metrics in FuncDiv with this command:
names(FuncDiv_alpha_metrics)Some alpha diversity metrics, from the macroecological literature, are only appropriate for absolute counts of individuals. We will focus on this subset, which returns the same values regardless whether the taxa abundances are absolute or relative (the latter being appropriate for sequencing data):
metrics <- c("richness", "shannon_index", "berger_parker_dominance", "ENS_pie",
"heips_evenness", "pielous_evenness", "gini_simpson_index",
"simpsons_evenness", "inverse_simpson_index", "faiths_pd")These metrics were largely ported over from the scikit-bio Python package, and you can read the metric descriptions here.
The list contains functions for computing each metric - you can see the function details with a command like this:
FuncDiv_alpha_metrics$richness
# function (x)
# {
# length(which(x > 0))
# }You can compute contributional alpha diversity with this command (with the func_tab and abun_tab objects as input):
alpha_out <- alpha_div_contrib(metrics = metrics,
func_tab = func_tab,
abun_tab = abun_tab,
in_tree = in_tree,
ncores = 1,
replace_NA = FALSE)Note: setting replace_NA = TRUE would replace all NA values with 0. This makes post-analyses easier, but would be misleading as generally NA values (for evenness for instance) occur when there are no contributing taxa present and so 0 is not actually correct.
Alternatively, if you have a data in contributional format you can run the same command with slightly different inputs (see below). Note that the contrib_tab is specified instead of the two tables. Also, the column names for each data type need to be specified. The output should be identical regardless of which approach is used.
alpha_out <- alpha_div_contrib(metrics = metrics,
contrib_tab = contrib_tab,
in_tree = in_tree,
ncores = 1,
replace_NA = FALSE,
samp_colname = "samp",
func_colname = "func",
taxon_colname = "tax",
abun_colname = "tax_abun")alpha_out is a list containing each respective table of contributional alpha diversity values. Each element is named by metric:
names(alpha_out)
# [1] "faiths_pd" "richness" "shannon_index" "berger_parker_dominance"
# [5] "ENS_pie" "heips_evenness" "pielous_evenness" "gini_simpson_index"
# [9] "simpsons_evenness" "inverse_simpson_index" Each table contains the values for each function and sample combination. For example, here is a subset of the observed richness output table.
alpha_out$richness[1:5, 1:20]
# ERR1190790 ERR1190797 ERR1190798 ERR1190800 ERR1190806 ERR1190809 ERR1190812 ERR1190814 ERR1190815 ERR1190816
# K00228 0 0 0 0 0 0 0 0 0 0
# K00231 0 0 0 0 0 1 0 0 0 0
# K00247 0 0 0 0 0 0 0 0 0 0
# K01474 0 0 0 0 0 0 0 0 0 0
# K01591 8 26 15 33 17 35 25 19 16 16
# ERR1190823 ERR1190828 ERR1190875 ERR1190881 ERR1190885 ERR1190886 ERR1190892 ERR1190895 ERR1190899 ERR1190900
# K00228 0 0 1 0 0 0 0 0 0 0
# K00231 1 0 0 0 0 0 1 1 1 1
# K00247 0 0 1 0 1 0 0 0 0 0
# K01474 0 0 0 0 0 0 0 0 0 0
# K01591 37 10 34 16 32 29 29 29 41 27Note that in this example the samples in the input taxa abundance were not rarified or otherwise controlled for differential depth between samples. This means that there are major differences in contributional diversity metrics between samples, which could reflect technical variation. For instance, the mean contributional richness values per sample vary substantially:
colMeans(alpha_out$richness)
# ERR1190790 ERR1190797 ERR1190798 ERR1190800 ERR1190806 ERR1190809 ERR1190812 ERR1190814 ERR1190815 ERR1190816 ERR1190823 ERR1190828
# 2.04 5.74 3.74 7.32 4.06 7.46 5.66 4.28 3.72 3.74 7.86 2.66
# ERR1190875 ERR1190881 ERR1190885 ERR1190886 ERR1190892 ERR1190895 ERR1190899 ERR1190900 ERR1190901 ERR1190905 ERR1190911 ERR1190912
# 7.66 3.32 6.74 6.46 6.64 6.16 8.92 6.26 5.80 3.12 6.90 7.40
...So, for subsequent analyses it would be important to correct for variation in total diversity. Mean-centring and scaling by the standard deviation by sample is one possible solution:
alpha_out_scaled <- lapply(alpha_out,
function(X) { data.frame(scale(x = X,
center = TRUE,
scale = TRUE)) })There are many ways these data could be analyzed. One possible analysis would be to make a heatmap of the data, which is easy to do with the ComplexHeatmap package. See below for a minimal example, displaying the transformed observed richness per function and sample combination.
library(ComplexHeatmap)
Heatmap(as.matrix(alpha_out_scaled$richness))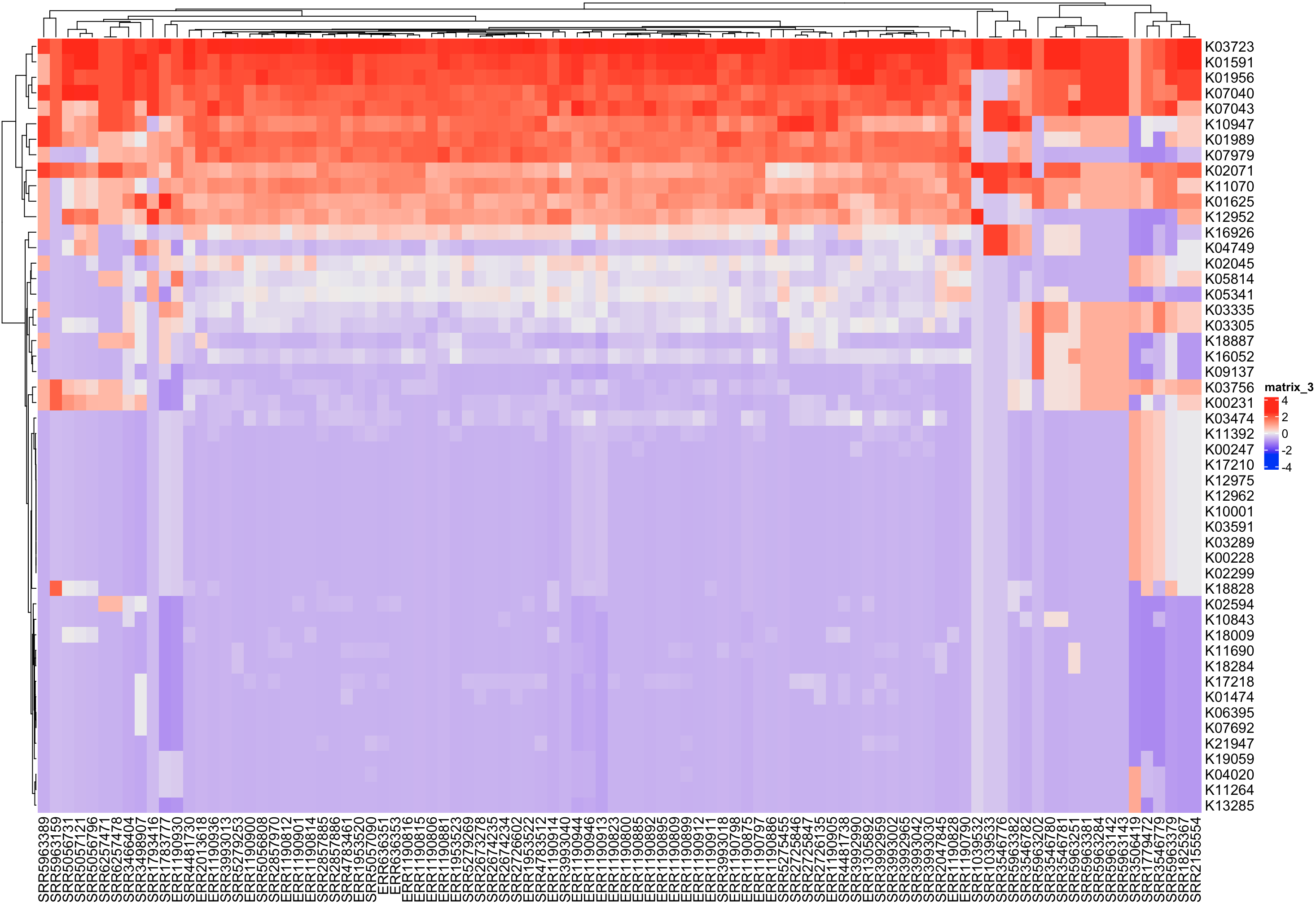
Beta diversity metrics restricted to the subsets of taxa encoding a particular function per sample can also be computed with FuncDiv. This will result in an inter-sample distance/divergence matrix for every function. This can be a little unwieldy, but can be useful in certain circumstances. In many cases it would be inefficient to return all these matrices within a single list. This can still be done (if return_objects = TRUE), but generally it is better to write the distance matrices directly to the disk as they are made (which is done by setting write_outfiles = TRUE below). This approach greatly reduces the memory burden and the output files can be found in plain-text format in specified output directory (outdir below).
Currently beta diversity metrics are restricted to those available through the parallelDist R package. You can see the metric types by typing ?parallelDist::parDist.
In this example, we will compute basic Jaccard distance ('binary') and Bray-Curtis distance ('bray'), which can be done with this command:
beta_div_contrib(metrics = c('binary', 'bray'),
func_tab = func_tab,
abun_tab = abun_tab,
ncores = 1,
write_outfiles = TRUE,
outdir = '/path/to/output/directory/')The above command will create a new directory with the specified name, and subdirectories for each distance metric specified. Within these folders there will be one tab-delimited file (named for each respective function), containing the distance matrix for the taxa encoding that function between samples. Sample ids are specified by the column names, which are also the same order as the rows (which are unnamed).