md-highlighter is a simple extension to highlight the most various file types about molecular modeling and molecular dynamics simulation. This extension provides comprehensive coverage of commonly used file formats under various popular tools, including Gromacs, Amber, AmberTools, NAMD, VMD, and more. It also performs detailed keyword parsing, helping users to locate and edit information within files more quickly. Users can even identify syntax errors quickly based on different display colors. Feel free to download and try it out, and don't hesitate to recommend it to your colleagues!
Supported format:
- NAMD: rtf, pdb, prm, psf, str, inp
- Amber: in, prmtop, inpcrd, prepin, frcmod, lib/off, ac, mc
- Gromacs: gro, atp, arn, rtp, hdb, r2b, tdb, mtp (pmx), ...
- small molecule: sdf, mol2 (modified from gromacs helper), ...
- PLUMED: .plumed.dat
For gromacs .top/.itp files, you may also install gromacs-helper. For VMD, install TCL. For Gaussian, install Gaussian Input File (gjf). But maybe I make my own syntaxes.
Tested in theme "Atom One Light".
Install to explore more file types!
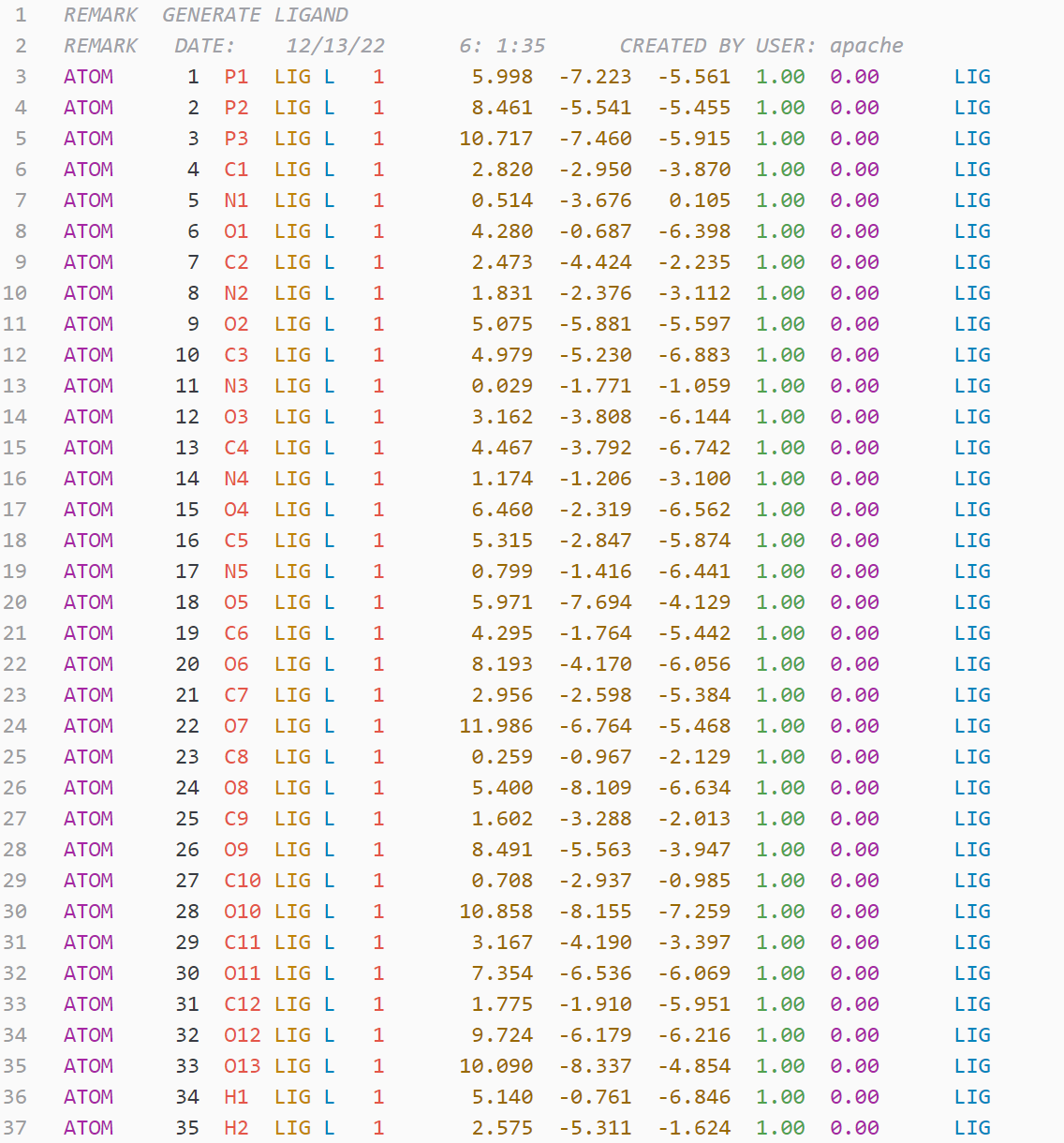

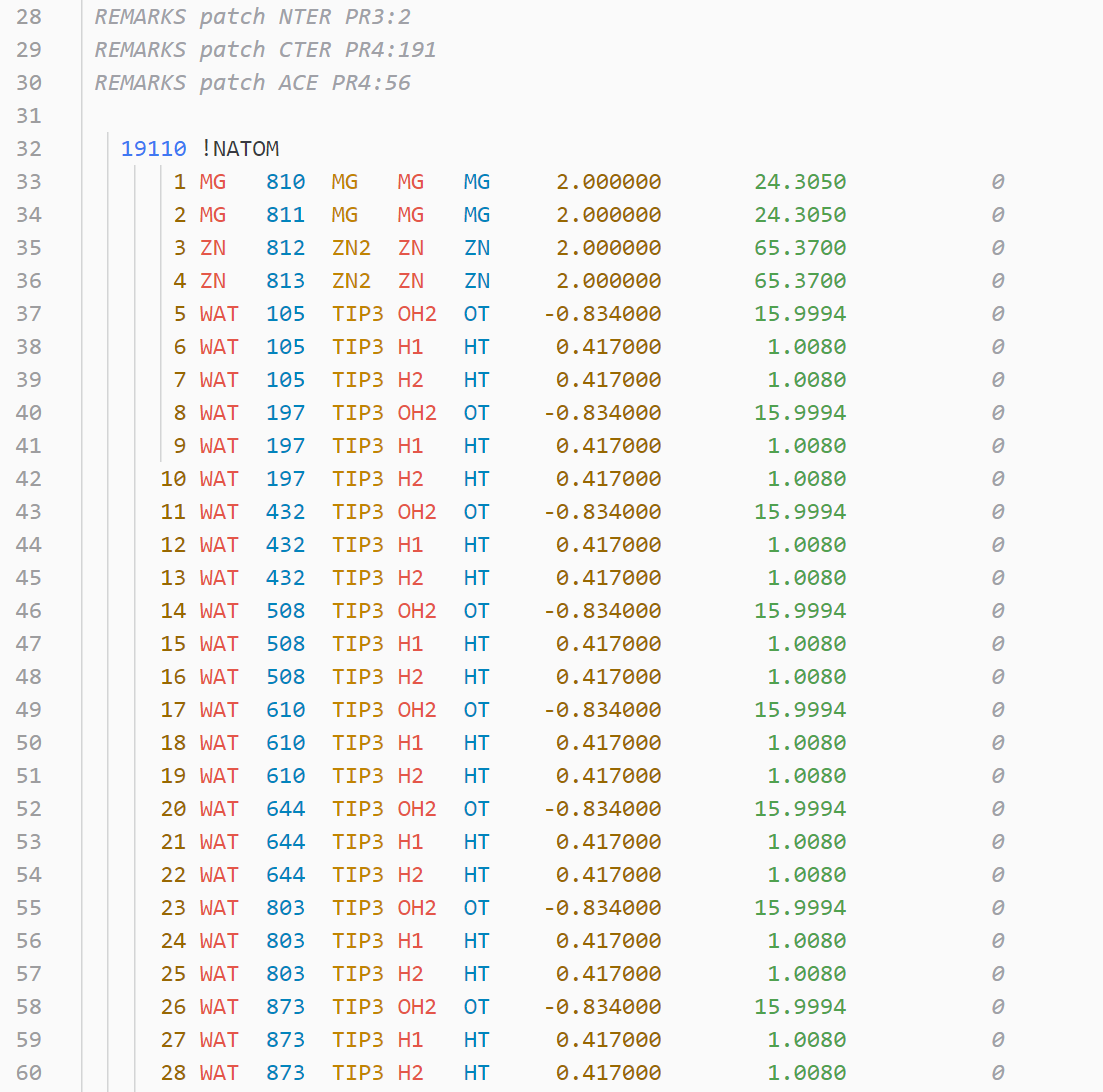
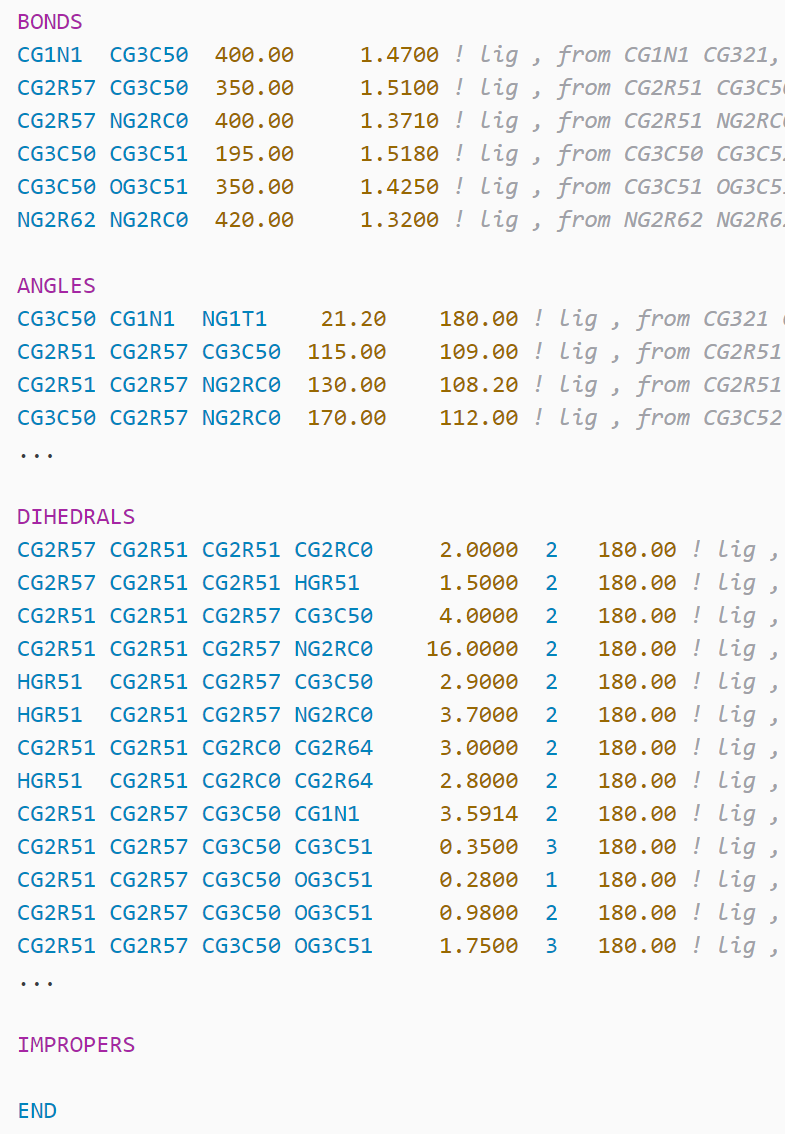
- variable.other.residue-number (except psf,gro)
- entity.name.type.residue-name (except hdb)
- constant.numeric.atom-number (except gro,lib)
- entity.name.tag.atom-name
- support.type.atom-type
- constant.numeric.mass
- constant.numeric.coordinate
- string.quoted.charge (rtf,psf,mol2,prepi, except rtf)
- string.quoted.element-symbol
- string.quoted.bond-number
- string.quoted.velocity
- support.type.segment-name (except psf)
- entity.name.function: a different kind of blue
But different file types might rendered with different colors...
- Are the files too colorful?
- PDB: CONNECT and TER highlight later terms
- RTF: the structure depiction after ! is italic...
- atom names
- with
',+,-in them are matched by\\S+but not[A-Z+-'] - not always start with numbers (
-?\\+?[A-Z]+[0-9]*[A-Z0-9_]*) - MTP: the last word in rotations
- with
- Amber .in files include various types; so do Gromacs .itp file
- PRMTOP: E-01, normal
constant? - FRCMOD: I know atomtypes can only be 1/2 characters long. Matching with this may solve the mismatching problem
- LIB: later section not read and implemented
- MTP/RTP: cannot match the last atom name?
- not fully tested files for OPLS series and more force field, where naming conventions might be different
TODO:
- add self-defined colors for aminoacids types (polar, nonpolar, etc.)
- add self-defined colors like pink, etc.
- sdf file?
This extension is created with the help of ChatGPT and New Bing in the beginning.
see CHANGELOG
Welcome to contact me for any problems: gxf1212@zju.edu.cn
Enjoy!