scChat: A Large Language Model-Powered Co-Pilot for Contextualized Single-Cell RNA Sequencing Analysis
Welcome to the scChat page. scChat is a pioneering AI assistant designed to enhance single-cell RNA sequencing (scRNA-seq) analysis by incorporating research context into the workflow. Powered by a large language model (LLM), scChat goes beyond standard tasks like cell annotation by offering advanced capabilities such as research context-based experimental analysis, hypothesis validation, and suggestions for future experiments.
Watch the demo of scChat in action below:
{kind=link}
If you found this work useful, please cite this preprint as:
@misc{lu2024scchat,
title={scChat: A Large Language Model-Powered Co-Pilot for Contextualized Single-Cell RNA Sequencing Analysis},
author={Yen-Chun Lu and Ashley Varghese and Rahul Nahar and Hao Chen and Kunming Shao and Xiaoping Bao and Can Li},
year={2024},
eprint={2024.10.01.616063},
archivePrefix={bioRxiv},
doi={10.1101/2024.10.01.616063}
}Data-driven methods such as unsupervised and supervised learning are essential tools in single-cell RNA sequencing (scRNA-seq) analysis. However, these methods often lack the ability to incorporate research context, which can lead to overlooked insights. scChat addresses this by integrating contextualized conversation with data analysis to provide a deeper understanding of experimental results. It supports the exploration of research hypotheses and generates actionable insights for future experiments.
Please read our scChat paper for more motivation and details about how the scChat works.
Model: scChat currently supports analysis using AnnData-formatted single-cell RNA sequencing datasets.
Capabilities: scChat integrates an LLM with specialized tools to enable tasks such as marker gene identification, UMAP clustering, and custom literature searches, all through conversational interactions.
To set up the project environment and run the server, follow these steps:
- Install the required dependencies:
pip3 install -r requirements.txt
Follow these steps to utilize the application effectively:
- type and enter export OPENAI_API_KEY='your_openai_api_key' in your terminal
- run python3 manage.py runserver
-
Open your web browser and navigate to:
http://127.0.0.1:8000/schatbot -
Recommended: Use Google Chrome for the best experience.
-
Periodically clearing the cache is recommended to ensure a smooth experience:
- Right-click on the page and select Inspect.
- Go to the Application tab.
- Under Cookies, remove
sessionid.
This will prevent previous chat sessions from being reprocessed.
- Upload scRNA-seq adata file (.h5ad)
- Upload sample mapping (.json file) (if required).
- Upload research context (.txt file) (if required).
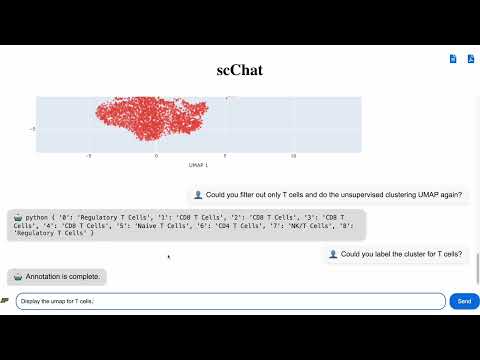
- Request to generate UMAP for overall cells in the scRNA-seq Analysis. ("Generate UMAP")
- (4) Will return a Python dictionary. You can then request to label/annotate clusters for overall cells. ("Label clusters for overall cells")
- You can now ask to display the annotated UMAP for overall cells or view the unannotated UMAP for overall cells.
- You can ask for rationale or research questions specific to your dataset.
- If needed, you can filter and process a specific cell type for detailed subtype clustering and annotation. ("Process <cell_type> cells")
- (8) would return a Python dictionary. You can then label/annotate clusters for the processed cell. ("Label clusters for <cell_type> cells")
- You can calculate cell population changes for overall cells or specific cell types. ("Calculate cell population change for <cell_type> cells")
- You can display the cell population change for previously calculated cell types. ("Display cell population change for <cell_type> cells")
- You can ask for reasoning, possible hypotheses, experimental designs, and additional insights.
- You can compare differential gene expression between two samples for a specific cell type. ("Calculate sample differential expression genes comparison for <cell_type> <sample_1> <sample_2>")
The datasets used for testing can be found at https://docs.google.com/spreadsheets/d/1NwN5GydHn0B3-W0DLcAfvnNtZVJEMUgBW9YyzXnS83A/edit?usp=sharing
The scChat_RAG component allows users to specify the organism, tissue type, and condition (normal or disease) for their single-cell RNA sequencing (scRNA-seq) analysis. The data is structured into human and mouse categories, with each tissue having two available conditions: normal and normal_and_cancer.
To run a scRNA-seq analysis with scChat RAG, users can configure the .json file by specifying the organism (human or mouse), the tissue type, and the condition (either normal or normal_and_cancer).
For example, to analyze normal blood cells from a human, the configuration would look like this:
{
"marker": "human",
"tissue": [
"blood"
],
"condition": "normal"
}This configuration will retrieve and process the relevant dataset based on the selected organism, tissue, and condition, enabling a customized and context-specific analysis.
- Adipose Tissue
- Blood
- Bone Marrow
- Brain
- Breast
- Eye
- Heart
- Intestine
- Kidney
- Liver
- Lung
- Ovary
- Pancreas
- Salivary Gland
- Skin
- Testis
- Adipose Tissue
- Blood
- Bone Marrow
- Brain
- Heart
- Kidney
- Liver
- Lung
- Ovary
- Pancreas
- Spleen
- Testis
Each tissue has a corresponding normal.json and normal_and_cancer.json file for both organisms, making it easy to switch between healthy and diseased conditions.
The scChat_RAG files were generated using data from:
CellMarker: a manually curated resource of cell markers in human and mouse
Published in Nucleic Acids Research, 2018
DOI: 10.1093/nar/gky900