Releases: merenlab/anvio
anvi'o v8, "marie"
We are happy to announce anvi'o v8 with the code name, "marie"!
After about 4,200 changes that introduced over 36,000 new lines of code, this stable release of anvi'o represents significant advancements over v7, and introduces many new features for integrated studies of microbial metabolism, genomic inversions, phylogeography of proteins, performance improvements, and fixes for known bugs.
This page intends to give you a summary of some of the notable changes that come with marie.

The code name recognizes Marie Tharp, an American geologist and oceanographic cartographer, who has made immense contributions to earth sciences. Marie was a pioneer in our understanding of oceans as she created the first map of the Atlantic seafloor with her colleague Bruce Heezen [1]. Her work showed that the bottom of our oceans were not only flat sediments but were also covered with canyons, ridges, and mountain ranges that spanned over 65,000 kilometers around the globe. Marie's revolutionary work emerged from her interpretation of data she was not allowed to collect since women were not allowed to be on ships during the 1950s. Marie compiled her physiographic diagrams from the data Bruce Heezen were able to collect [2]. She did not step on a ship until 1968, and the early evidence she had for seafloor features was initially dismissed as 'girl talk' [3].
[1] https://en.wikipedia.org/wiki/Marie_Tharp
[2] https://www.lyellcollection.org/doi/abs/10.1144/GSL.SP.2002.192.01.11
[3] https://www.youtube.com/watch?v=gsQGOJtwdv0
The code name was a suggestion by Zena Cardman, a Marine Microbiologist and a NASA Astronaut. The release notes were written by Meren, Iva Veseli, and Matt Schechter, who are among the developers of anvi'o. The notes were proofread by Katy Lambert-Slosarska, who is a MSc student at the International Max Planck Research School of Marine Microbiology (MarMic).
New anvi'o programs, artifacts, and workflows
The new version of anvi'o comes with a few new programs:
- anvi-compute-functional-enrichment-across-genomes
- anvi-compute-functional-enrichment-in-pan
- anvi-compute-metabolic-enrichment
- anvi-delete-functions
- anvi-display-functions
- anvi-get-codon-usage-bias
- anvi-get-metabolic-model-file
- anvi-get-pn-ps-ratio
- anvi-get-tlen-dist-from-bam
- anvi-merge-trnaseq
- anvi-plot-trnaseq
- anvi-profile-blitz
- anvi-reaction-network
- anvi-report-inversions
- anvi-run-cazymes
- anvi-search-palindromes
- anvi-search-primers
- anvi-search-sequence-motifs
- anvi-setup-cazymes
- anvi-setup-kegg-data
- anvi-setup-modelseed-database
- anvi-setup-user-modules
- anvi-summarize-blitz
- anvi-tabulate-trnaseq
- anvi-script-as-markdown
- anvi-script-compute-bayesian-pan-core
- anvi-script-estimate-metabolic-independence
- anvi-script-filter-hmm-hits-table
- anvi-script-gen-function-matrix-across-genomes
- anvi-script-gen-functions-per-group-stats-output
- anvi-script-gen-genomes-file
- anvi-script-gen-user-module-file
- anvi-script-permute-trnaseq-seeds
And a few new artifacts:
- bam-stats-txt
- bams-and-profiles-txt
- cazyme-data
- contig-inspection
- dna-sequence
- enzymes-list-for-module
- enzymes-txt
- external-structures
- functions-across-genomes-txt
- gene-cluster-inspection
- hmm-hits-across-genomes-txt
- hmm-list
- inversions-txt
- markdown-txt
- metabolic-independence-score
- modifications-txt
- paired-end-fastq
- palindromes-txt
- primers-txt
- quick-summary
- reaction-network
- reaction-network-json
- reaction-ref-data
- seeds-non-specific-txt
- seeds-specific-txt
- trnaseq-contigs-db
- trnaseq-plot
- trnaseq-profile-db
- trnaseq-seed-txt
- user-metabolism
- user-modules-data
- variability-profile-xml
In addition, this release makes available three new Snakemake workflows that are accessible via the anvi'o program anvi-run-workflow: trnaseq, ecophylo, and sra_download.
A new subsystem for metabolic modeling
One of the biggest news in this release is the set of programs now anvi'o includes for metabolic modeling. These programs are emerging as a by-product of collaborative projects in C-CoMP, or the Center for Chemical Currencies of a Microbial Planet, and under the leadership of Samuel Miller.
Using the integrated anvi'o metabolic modeling subsystem, one can generate a biochemical reaction network suitable for metabolic modeling from the annotations in a genome or a pangenome using the new program anvi-reaction-network. This works on both individual genomes (using a contigs-db and pangenomes (using a genomes-storage-db). The resulting network is stored in corresponding anvi'o database for programmatic access, and can be exported into a JSON file for inspection and downstream usage (i.e., as input into a program for flux-balance analysis) via another new program, anvi-get-metabolic-model-file.
These programs rely on KEGG Orthology (KO) annotations of protein-coding genes and reference data in the [ModelSEED ...
v7.1
A minor release with many bug fixes and new anvi'o toys.
Please see our up-to-date installation instructions at https://merenlab.org/install-anvio.
anvi'o v7, "hope"
We are happy to announce anvi'o v7 with the code name, "hope"!
After more than 3,000 changes that introduced about 35,000 new lines of code, this stable release of anvi'o represents one of the largest leaps forward in the history of the platform that introduces many new features, performance improvements, and fixes for known bugs.
This page intends to give you a summary of some of the notable changes that come with hope.

The code name recognizes Hope E. Hopps as a tribute to all laboratory technicians whose contributions have often been poorly recognized in science. This is despite the fact that technicians not only ensure accuracy, efficiency, and reproducibility in any laboratory, but also push the boundaries of science as much as any other member of their groups, if not more in many cases. Hopps was a specialist in infectious diseases and in 1966 she developed, together with Harry M. Meyer and Paul J. Parkman, a highly effective vaccine for rubella, a viral infection which caused more than 30,000 stillbirths in the United States alone between 1962 and 1965. Despite her role in the vaccine development, in a historical photograph by the NIH that portrays the rubella vaccine development team, Hopps was only identified as "Female Lab Technician" until recently, even though the caption of the same photograph explicitly named Meyer and Parkman. The unfair treatment of laboratory technicians remains to be commonplace in today's science. In fact, "not more than a technician's job" can serve as an argument for professors when they wish to refuse the recognition of one's contributions to science. We can't ignore the significant progress we have made as a community during the past few years. But while we continue working on increasing the diversity, equity, and inclusion in science, we must also recognize and face the implicit and explicit biases against those in science who are not PIs, post-docs, or graduate students.
Disclosures: The code name was a suggestion by Alon Shaiber, a Genomics Data Scientist at Weill Cornell Medicine. The release notes were written by Meren and proofread by Iva Veseli. Alon, Meren, and Iva are among the developers of anvi'o.
New help pages for anvi'o programs and artifacts
As anvi'o developers, we always knew the critical importance of providing our users with extensive tutorials so they can find their way through their data themselves. However, as anvi'o matured, the number of anvi'o programs and artifacts increased dramatically. This created a bottleneck since every anvi'o tutorial assumed that our users knew about the common concepts in anvi'o (such as 'the profile database' or 'a collection') or common anvi'o programs (such as 'anvi-profile' or 'anvi-interactive'). Solving this fundamental problem required us to think of an entirely new technical approach to our documentation that is now in place.
We have now implemented a system (#1425) that makes two things possible:
- (1) For anvi'o developers, a means to quickly describe their contributions without leaving the environment where they write code (for instance, here is the description of 'collection' in the codebase),
- (2) For anvi'o users, a means to be able to see that information on a web page where all anvi'o programs and concepts are interconnected (for instance, here is the description of 'collection' on the web page).
This way, anvi'o could accumulate information from its developers without burdening them and present it to its users in a way where self-learning is possible. However, there was one significant problem: retrospectively describing all the things that have already been implemented in the codebase. Enter Jessica Pan (@Jessica-Pan), an undergraduate student at the MIT. Jessica took the responsibility of describing existing anvi'o programs and artifacts a few months ago and with the guidance of other anvi'o developers, Jessica was able to populate this technical framework with her words and descriptions (#1470), which added more than 200 files and tens of thousands of words of documentation to the codebase.
With this release, we are happy to also release the first outputs of this documentation project here, with the hope that it will make your life with anvi'o a little easier going forward:
Perhaps it will not be a surprise to the long-term anvi'o users that this documentation system is also connected to our command-line programs. Thanks to this, they will be able to offer you more useful help menu outputs. For instance, if you were to type anvi-interactive --help in your terminal in v7, you would see the following section at the end of the help menu, so you can click on the link to go to the online description of the program and browse through examples and artifacts associated with it:

If you visit the help pages you will see there are 'edit' links under every file. It is our way of inviting the rest of the community to contribute to these pages with their own experiences with anvi'o tools. If you have ideas to make it better, come to our Slack channel for a discussion, or file a GitHub issue. We are all ears.
Significant performance improvements
This version will likely be remembered for significant performance improvements by multiple heroes, including Evan Kiefl (@ekiefl), Iva Veseli (@ivagljiva), and Ryan Moore (@mooreryan). Here is a glimpse of what happened in v7 compared to v6:
-
Profiling BAM files is one of the most critical steps in anvi'o and the program anvi-profile has been a nightmare for memory and processing time. Thanks to @ekiefl's significant improvements that influenced the runtime and memory requirements of the profiling step (#1362, #1339),
anvi-profileis now ~17 times faster. -
One of the first steps in any anvi'o workflow is turning boring FASTA files into talented contigs databases that can be used by many anvi'o programs. Yet, the program anvi-gen-contigs-database has not been multithreaded, which has been a significant performance bottleneck before (#1344, #1431). Responding to our plea on Twitter, @mooreryan has made remarkable contributions to the anvi'o codebase (while wrapping up his PhD, that is) (#1437, #1468, and #1445). As a result, anvi-gen-contigs-database is now multithreaded, at least two times faster than before, and can take advantage of your fancy clusters to be even more efficient.
-
After making our users who care about the quality of their MAGs go through so much pain and suffering for years, anvi-refine in this release is ~13 times faster after significant improvements in its memory requirements (#1455, #1458), thanks to help from Xabier Vázquez-Campos (@xvazquezc).
-
Our dealings with HMMs also benefited from major performance improvements. Thanks to @ivagljiva's efforts (#1413), the two frequently used programs, anvi-run-hmms and anvi-run-pfams that rely on HMMER, will perform HMM annotations ~3x faster.
An integrated subsystem for metabolic reconstruction
We are also thrilled to announce that starting with this release, anvi'o includes a new suite of programs for predicting metabolic capabilities for genomic and metagenomic data, thanks to @ivagljiva's extensive work that started with #1413. The new programs in this release rely on the extensive set of resources in the Kyoto Encyclopedia of Genes and Genomes (KEGG) for gene annotation and metabolism estimation, although in future releases we will expand source resources for metabolic reconstruction.
As a part of this subsystem, this release introduces a new database, the anvi'o MODULES.db, which is generated from parsed KEGG data files (such as KOfam HMM profiles and text-based descriptions of KEGG MODULE) and used by subsequent programs detailed below for easy, organized access to metabolic data. A version tracking system ensures metabolism estimation is run using the same [MODULES.db](https://merenlab.org/software/anvio/help/main/artifacts/mo...
v6.2
v6.1
anvi'o v6, "esther"
We are happy to announce a new version of anvi'o, "esther" (easy install through conda, quick try via docker)).
After nearly 9,000 changes that introduced about 16,000 new lines of code, the current version of anvi'o represents many fixes to big and small bugs, as well as new features. This page intends to give you a summary of most notable changes that come with esther.

The codename is a small tribute to Esther Lederberg (1922-2006), an American microbiologist who studied plasmids and bacterial viruses. Lederberg discovered lambda phage, an E. coli virus that is commonly used in bacterial genetics and molecular biology to deliver DNA into a recipient organism. This led to her description of specialized transduction, that occurs when a prophage improperly excises from the host chromosome carrying host DNA in addition to the viral DNA. In collaboration with her husband, Lederberg developed the technique known as replica plating, which allows repeatable inoculation of bacterial colonies. Lederberg and Luigi L. Cavalli-Sforza discovered the Fertility factor or F-plasmid in E. coli. This is a sequence of DNA that lets the host cell transfer genetic material via a rod-like structure into recipient cells (conjugation). Despite her many incredible scientific accomplishments, she was constantly overshadowed by her husband. She was not appointed to a tenured position while they were both faculty at Stanford, and after their divorce she had a difficult time retaining her appointment. We dedicate anvi'o version 6 to the memory and revolutionary discoveries of Dr. Lederberg.
Real-time estimation of genome taxonomy
Working with genomes often requires insights into their taxonomy. This becomes a critical need especially in genome-resolved metagenomics studies as we are burning to find out where the genomes we reconstruct from metagenomes fit in the tree of life. Until this esther, anvi’o did not offer anything to address this need, however, this new version comes with a novel solution that covers both the interactive interface during binning:

and the terminal environment to survey existing collections of genomes:
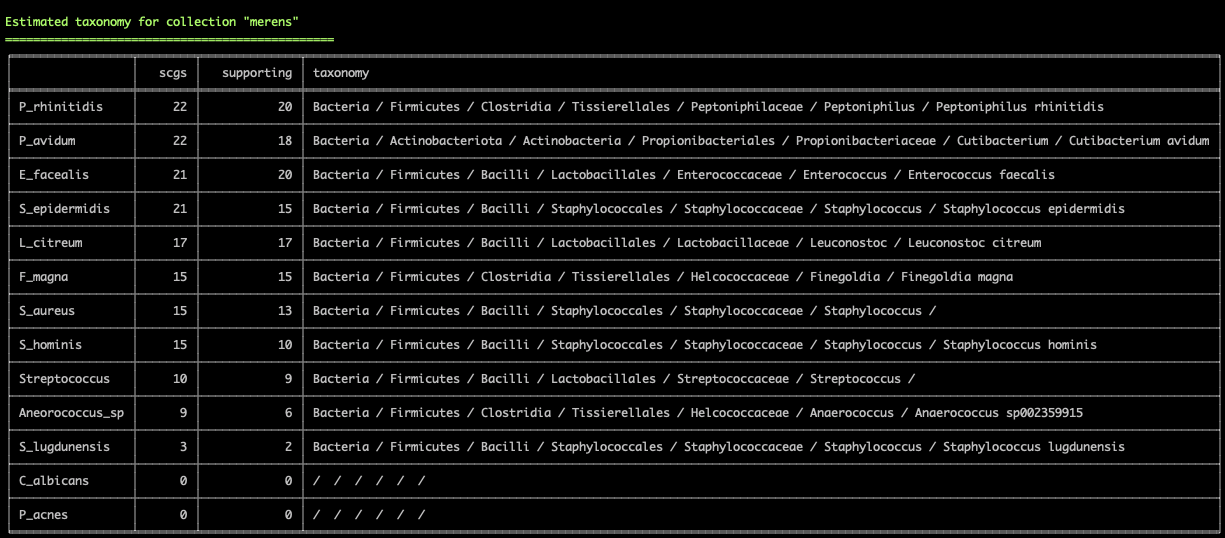
These two examples are from the infant gut dataset by Sharon et al (2013), which we often use to demonstrate anvi'o features, but we can't wait to hear from you to learn about your experience with this feature.
Please read in this article the usage details, our thanks to The Genome Taxonomy Database for making their raw data public, and potential caveats of our approach:
http://merenlab.org/scg-taxonomy
None of this would have been possible without the coding help from Quentin Clayssen and Özcan Esen, and critical suggestions from Alon Shaiber.
A new tool for genome de-replication
De-replication is a critical need to minimize bias in metagenomic read recruitment analyses. In our previous studies we had performed de-replication with a series of Python scripts, but no more. Thanks to Mahmoud Yousef and Evan Kiefl's efforts, we now have two new programs, anvi-compute-genome-similarity and anvi-dereplicate-genomes, integrated with metagenomic and pangenomic workflows in anvi'o and use sourmash and PyANI in the backend.
A tutorial for their usage is on the way!
Support for more binning algorithms
In previous versions of anvi'o we had a native module for CONCOCT, one of the popular binning algorithms for automatic clustering of contigs into genome bins. We have changed that behavior in this version. You will still be able to use the program anvi-import-collection to import binning results from ANY binning software as before, but anvi'o will also be able to automatically use binning tools existing on your system through our new program anvi-cluster-contigs. Here is a command line output to give you a sense of it:

This framework is highly modular, so the integration of new binning algorithms is extremely straightforward thanks to Özcan Esen's excellent design. If you are a programmer you can take a look at the module for MaxBin2 or BinSanity to develop one for your algorithm for benchmarking or testing efforts.
Effective ways to inspect and visualize contig coverages
Recognizing the importance of actually 'looking' at data, we have been putting a lot of emphasis on the inspection capabilities of anvi'o. When it comes to metagenomic read recruitment and coverages, inspecting contigs can be critical to gain deeper insights into what is actually going on.
In this version we have two new programs. The first one is anvi-inspect. The inspect page of anvi’o is very useful for careful examination of contig coverages and single nucleotide variants. Sometimes this might even be all you want. This new program enables you to immediately pull up the inspection page of a given contig without going through the whole hassle of opening the interactive interface.
We often feel the need to put coverage patterns of contigs in presentations or publications. Yet it becomes challenging when there are too many samples in a dataset as it makes it harder to study or save patterns comfortably using the interface. So we thought it would have been very useful if anvi'o could export coverage statistics using ggplot, but we didn't know enough R to be able to do this properly. As a result, we did what anyone who wish to work with talented people would do --we asked for help on Twitter:

Our call for help was heard by Ryan Moore, who actually developed a new anvi'o program that did exaxtly what we thought you would need, and much more: anvi-script-visualize-split-coverages (we sent him an anvi'o t-shirt as a token of our deep gratitude for his contribution, but we never got a photo back, so we don't know whether he is wearing it).
This program can export split coverages along with single-nuleotide variants on them into PDF files for even very large numbers of samples. It uses the output files anvi'o generates through anvi-get-split-coverages and optionally anvi-gen-variability-profile. The output is customizable with respect to plot color, axes, SNV color and grouping of samples. The tutorial for this feature will soon be on our web page.
Improved genome completion/redundancy estimates
New single-copy core gene collections
Starting with this version, we no longer use Campbell et al. and Rinke et al. single-copy core gene (SCGs) HMM sets to estimate completion of bacterial and archaeal genomes. Instead, we are using a modified version of the bacterial single-copy core gene collections Mike Lee recently described, and a set of BUSCO HMMs Tom Delmont curated. Now anvi'o can estimate the completion of bacterial, archaeal, and protist genomes (#1150).
New random forest domain of life classifier
In previous versions anvi'o has relied on multiple heuristics to predict the domains of selected contigs or genomes for the determination of which SCG collection to use to estimate and display completion and redundancy. In this version we have a brand new random forest classifier to take care of this challenging task. This robust classifier with appropriate addition of noise solves this issue like magic, and when you have a bunch of genomes, it gives you proper estimates in the interface (the example is also from the infant gut dataset),
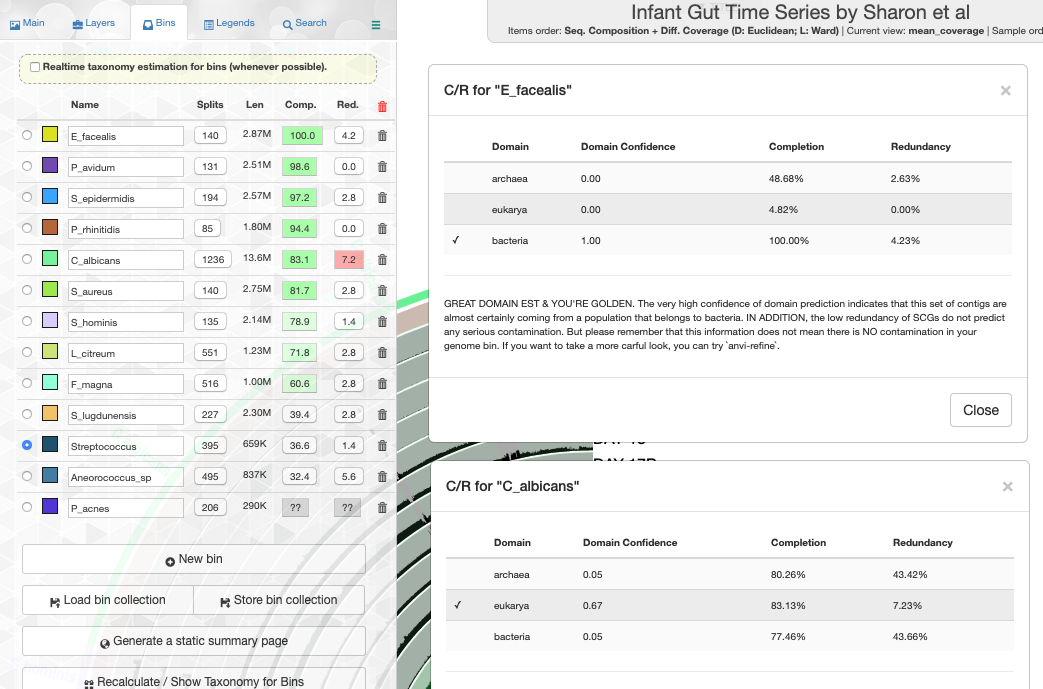
or in the terminal,

Undo/Redo for the interactive interface
Yes. This feature is finally here. Now when you make a mistake while curating or refining your genomes using anvi-interactive or anvi-refine, you will be able to use Ctrl + Z and Ctrl + Shift + Z key combinations for undo and redo your binning decisions. If you can't contain your e...
anvi'o v5, "margaret"
We are happy to announce a new version of anvi'o, "margaret".
After nearly 1,500 changes that introduced about 15,000 new lines to the anvi'o codebase and removed about 4,000 from it, the current version includes many fixes to big and small bugs, as well as new features. This page intends to give you a summary of most notable changes that comes with margaret.

The codename is a small tribute to Margaret Oakley Dayhoff, an American physical chemist, who is known as the founder of bioinformatics. Dayhoff developed first programmable computer methods to compare protein sequences, and published in 1965 a book titled "Atlas of Protein Sequences and Structure", which is considered as of today the first text book of bioinformatics. The codename was suggested by Mick Watson, and won the popular vote on Twitter. Dayhoff sadly died at an early age of 57 in 1994, shortly before bioinformatcis emerged as a distinct field. However, her astonishing contributions to life sciences, such as the development of essential approaches for protein sequence comparison and evolutionary tree construction, still constitute some of the most common approaches in our bioinformatics toolkit.
Your new disconcerting toy: GC-content overlaid on reference contexts
Metagenomic read recruitment often results in wavy coverage patterns in the reference context. This phenomenon, which can be attributed to three major sources, can result in up to an order of magnitude coverage difference for genes within the same contig. While we are kind enough to leave those alone who solely work with metagenomic short reads to quantify functions in metagenomes in their blissful world, we wanted to include in this version of anvi'o something so you can overlay GC-content change throughout your contigs to see whether variation you observe in the context of some of your key genes is largely driven by GC-content or not:

This is not yet anything but a qualitative insight for you to make sense of to what extent variation in coverage could be explained by deterministic factors that have nothing to do with the biology of your system given the metagenome, but it shows that more quantitative insights into this could be useful. We will think about this going forward, and we are open to your suggestions!
A new anvi'o workflow management system for serious anvians
This new version of anvi'o includes a new program anvi-run-workflow, which provides an interface to our new module that implements snakemake-based anvi'o workflows.
These workflows offer accessible, reproducible, and comprehensible solutions for complex analyses that may include hundreds of samples. We have been using anvi-run-workflow every day in our lab since it first appeared in our master repository, and we are happy to make its power available to you as soon as we could.

There will be an extensive tutorial very soon, but until then you can send your questions to Alon (smiley).
Single-codon variants for a more powerful framework to study microbial population genetics
Anvi'o already could make sense of single-amino acid variants (SAAVs) in environmental metagenomes. But working with SAAVs was limiting our ability to infer and quantify neutral processes that may not result in changes in the amino acid sequence. We changed our design in such a way, now anvi-profile can characterize single-codon variants (SCVs) if --profile-SCVs flag is declared. We updated our reference manual for variability analysis to include new sections describing SCVs and SAAVs.
With SNVs, SCVs, and SAAVs, anvi'o v5, deserving of its codename, offers a robust framework to investigate population genetics of environmental microbes, while SCVs and SAAVs leverage our ability to tease apart evolutionary forces acting upon them. We hope you enjoy these new toys, and feel free to get in touch with us if you have questions or suggestions.
Visualize environmental variation on protein structures through the new Structure DB
Our efforts to push the boundaries of investigations of environmental variation within microbial populations reaches to a new level in this release with a brand new ability about which we are very excited: linking variation to predicted protein structures.
With the new structure database associated workflows anvi’o can predict the tertiary structure of genes identified from a contigs database using the Protein Database Bank. Then, it can directly overlay onto the predicted protein structures the variability data from your metagenomes in the form of SCVs and SAAVs. All of this is accomplished in just two new programs, anvi-gen-structure-database and anvi-display-structure.
We believe that this nexus between structural biology and metagenomics will elevate environmental metagenomics into the realm of biophysics, and enable investigations into evolutionary processes driving the diversity of proteins that could not be learned from sequence analyses alone.
With these new advances come two new dependencies to additional open-source software, for which we are very grateful: MODELLER and DSSP.
Here is a teaser from the new interactive anvi-display-structure interface:

We will soon make available an extensive tutorial to describe this workflow in detail. Until the, you can send your questions to Evan and Ozcan.
Computing average nucleotide identity for genomes in pangenomes
This release also includes significant improvements for our comparative genomics and pangenomics workflows.
One of these improvements is the inclusion of a new program, anvi-compute-ani, to calculate the average nucleotide identities across a given set of genomes, which can be automatically added into any anvi'o pangenome.
For instance, this is an anvi'o pangenome of the 31 Prochlorococcus isolates we played with in our recent paper:
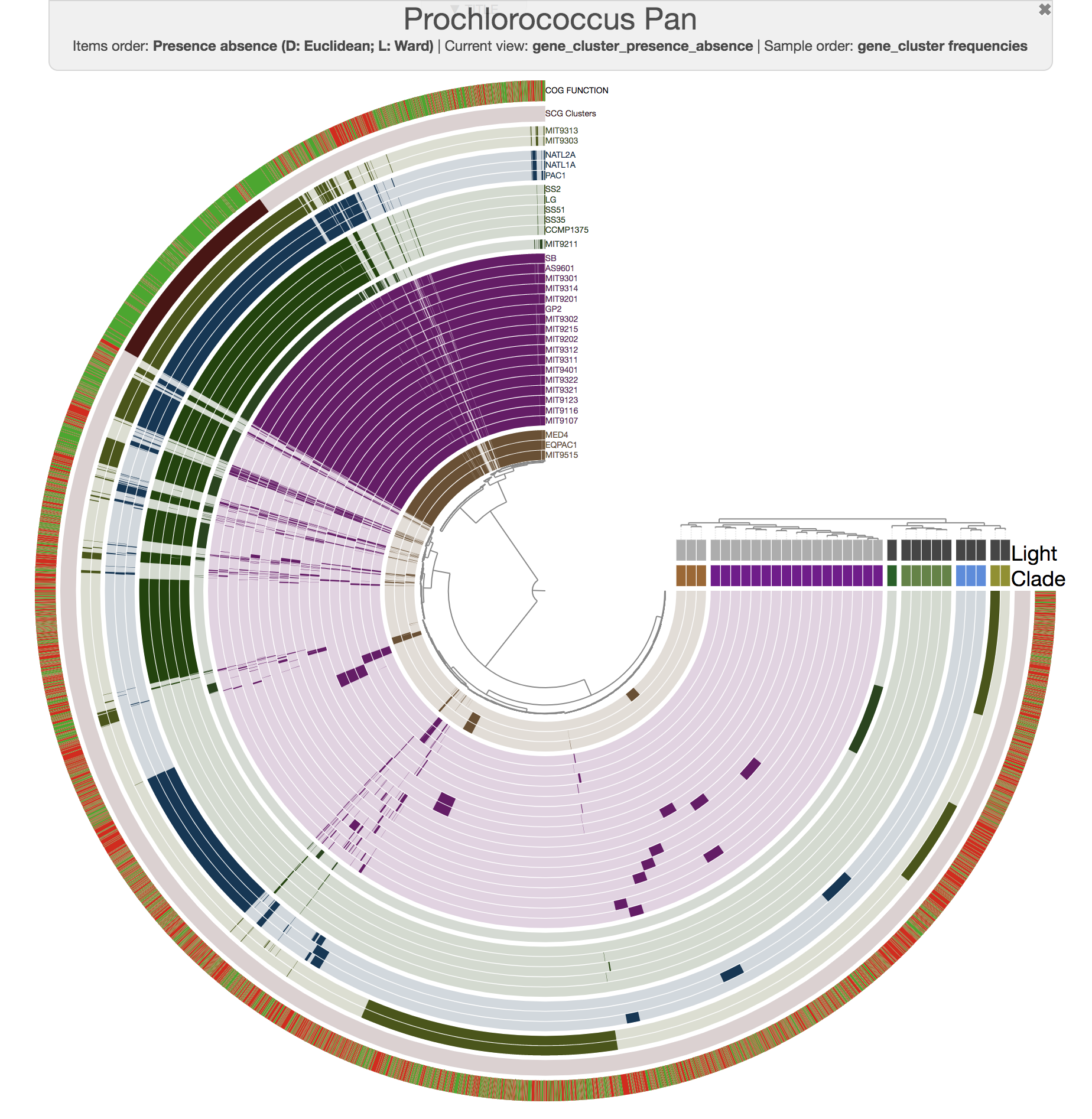
And this is what you get when you run anvi-compute-ani:
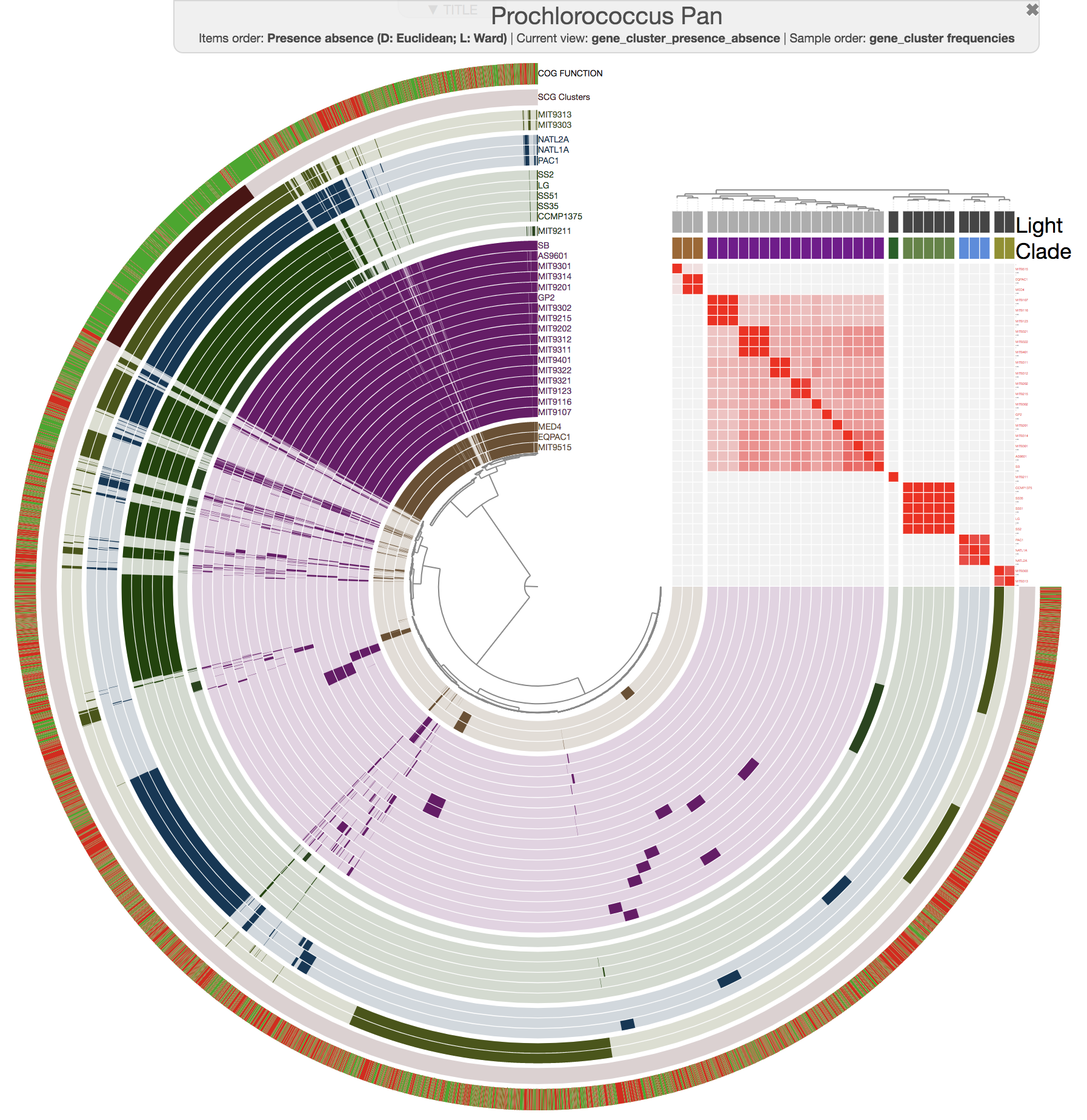
Mike Lee had suggested this as an option a long time ago. We are happy to finally deliver this functionality, which uses pyANI as a backend, for which we are thankful for its developers.
We updated our tutorial on pangenomics to describe intermediate steps.
A new approach to explore functional enrichment in pangenomes
This version of anvi'o also incluedes a new analytical framework to study functional enrichment in a given pangenome based on any arbitrary organizations of genomes. You simply define how would you like to partition your genomes, whether based on a phylogenetic tree or a dendrogram that anvi'o computed from gene cluster distributions, and this new tool finds functions that are enriched in those groups (i.e. functions that are characteristic of a given group of genomes, and predominantly absent from genomes from outside this group).
This is done by the new program anvi-get-enriched-functions-per-pan-group, and Alon extended our current tutorial on pangenomics with an extensive description of how it works.
Native functional annotation options += PFAMs
If you have your own functional annotations for your genes in an anvi'o contigs database, it is quite straightforward to import them via anvi-import-functions program. Anvi'o v3 had made available another program to automatize the annotation process, anvi-run-ncbi-cogs, if you were fine with NCBI's Cluster of Orthologus Groups. This release contains a new program, anvi-run-pfams to use the collection of HMMs produced by the European Bioinformatics Institute based on UniProt.
Tree modification through the interactive interface
It has been a challenge to deal with phylogenetic tree operations in anvi'o interactive interface. This version includes a significant code refactoring effort, which makes possible to have new toys that we could not have before. These new toys include basic tree editing and storage abilities such as re-rooting trees, rotating and collapsing branches. You can even see the branch support values in the mouse tab of the anvi'o interactive interface. These functions ar...
anvi'o v4, "rosalind"
We are happy to announce a new version of anvi'o, "rosalind".
After nearly 300 changes that introduced about 15,000 new lines, and removed about 7,500 from the anvi'o codebase, the current version includes many bug fixes, as well as some new features. This release note intends to give you a summary of most important changes.

The codename is a small tribute to Rosalind Franklin, the British biophysicist whose work, among other advances in life sciences, led to the discovery of the DNA double helix. This codename was inspired by Emily Crossette's suggestion, 'esther', "after Esther Lederberg, who co-developed a replica plating method with her husband but was largely unrecognized and discriminated against as a woman scientist". Emily explained that her suggestion was to "celebrate how far we have come as a scientific community and look to the future". Yes. We fortunately did not stay where we were, but we are still far from where we could have been. We remember these women and many others with respect and gratitude, and understand our responsibility to make sure the younger generations of scientists will not suffer from the kinds of discrimination to which their professors were subjected.
An elegant way to upgrade anvi'o databases
Upgrading anvi'o databases is now simpler than ever. With this change, the number of excuses you can use to not switch to the newest version of anvi'o goes from "0" to "-1". Just saying.
We now have a single program, anvi-migrate-db, that upgrades any anvi'o database to the latest version in one step.
As a part of this change we replaced all HDF5 files, which resulted in tremendous performance gain (especially in pangenomic operations that required access to the genome storage database), and up to 10-fold reduction in disk storage needs (for auxiliary data files). As a result, these changes did occur: No more CONTIGS.h5 --the content of this file is now a part of the CONTIGS.db (yay for less clutter). No more SAMPLES.db (more on this down below). Genome storage and auxiliary data files now have .db extensions rather than .h5 as they are now SQLite databases, instead of HD5 files.
Improvements in the pangenomic workflow
We made multiple very critical improvements in our pangenomic workflow. Here is a list of them:
These are the gene clusters you are looking for. Now it is possible to "select" gene clusters programmatically both from the command line, and from the anvi'o interactive interface through the combination of filters. We thank Ryan Bartelme for pushing us to improve our pangenomic workflow as he once again did in #668. Gene clusters that match to these filters are highlighted immediately on the interface, and can be added into any bin/collection for summary:

Search gene clusters by function. We also now have the capacity to search for gene clusters that describe genes with functions of interest through the command line as well as the interactive interface:
Parallel alignment. After identifying all gene clusters in a given pangenome, anvi'o by default would use muscle or famsa to store multiple sequence alignments for amino acid sequences in each gene cluster. This was one of the most time consuming steps of the pangenomic workflow. With v4, anvi'o uses as many cores as you wish anvi'o to use to parallelize amino acid alignments per gene cluster. It changes a lot.
Forced synteny. Gene clusters in a pangenome are by default organized based on their distribution across genomes (so that is the dendrogram in the center). However, with this version there are additional ways to order them, including ordering them by "synteny". In this forced organization you get to choose one of the genomes in your analysis from the "item orders" combo box, which tells anvi'o that you wish to order all gene clusters in your pangenome based on the order of genes in that genome. We found it to be an efficient way to study missing genomic loci, and other not-so-straightforward-to-spot phenomena.
Everything is better in color. Arguably, one of the most important improvements to the pangenomic workflow was the addition of an amino acid alignment conservancy coloring algorithm. This was done in #732 by Mahmoud Yousef, who is currently a second year Computer Science student at the University of Chicago. Mahmoud also very kindly wrote a blog post to explain the details of this algorithm with examples: http://merenlab.org/2018/02/13/color-coding-aa-alignments/.
Gene popups. Now you can click gene caller ids next to the amino acid sequence alignment in inspection pages, and enjoy these functional popups to access any information (#680):

Cleaner terminology. After consulting with the community, we changed all instances of 'protein clusters' in our pangenomic workflow with 'gene clusters'.
Metapangenomics: linking pangenomes and metagenomes
Anvi'o comes with powerful analytical tools to study pangenomes and metagenomes. Now you can take things one step further with the same ease-of-use.
We define metapangenomics as the outcome of the analysis of pangenomes in conjunction with the environment where the abundance and prevalence of gene clusters and genomes are recovered through shotgun metagenomes. This version includes a new program, anvi-meta-pan-genome, that brings the power of metapangenomics into a single command line. Please read our paper on the Prochlorococcus metapangenome to see how this concept could apply to your research.
Improvements in the interactive interface
This release also include multiple notable improvements in the interactive interface.
The 'max coverage' fix we all needed but didn't know. Inspection pages are great to investigate coverage data and single-nucleotide variants in a single-nucleotide resolution, however, it was not quite easy to make visual sense of data when coverage values dramatically differed between samples, or short but non-specific mapping pushed maximum coverage values too high to make sense of the actual population coverage in the context of long contigs. In v4 you will see additional buttons in the inspection pages to mitigate these kinds of visual imperfections. Here is some action for you skeptics:
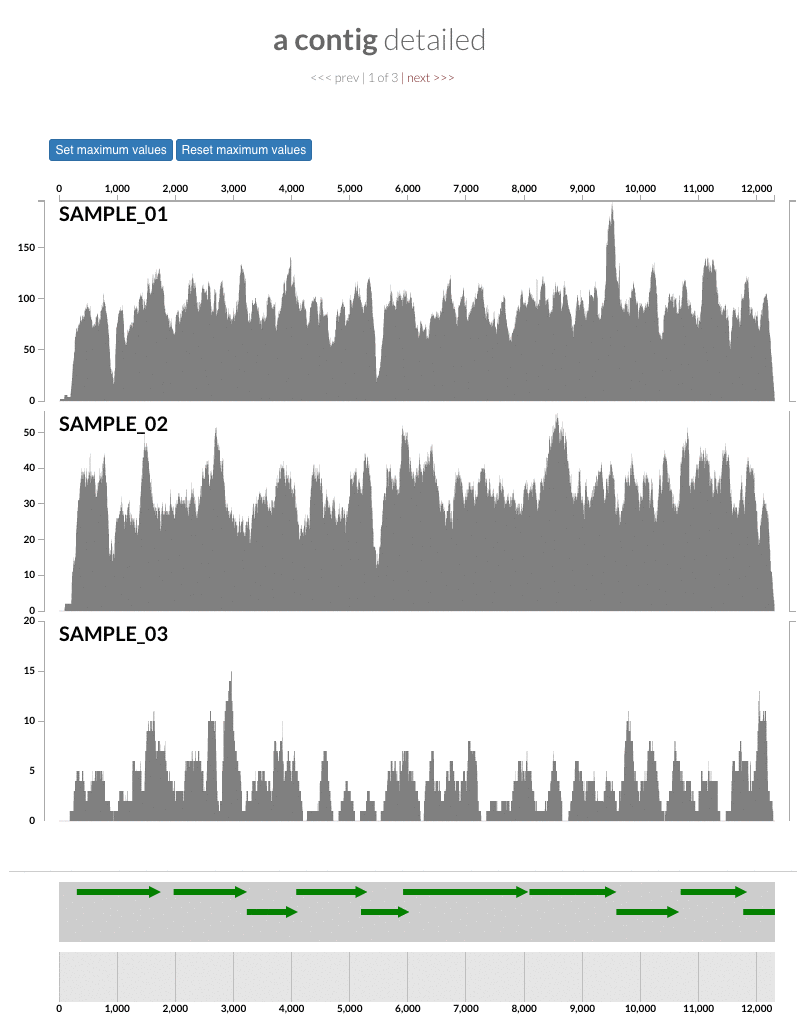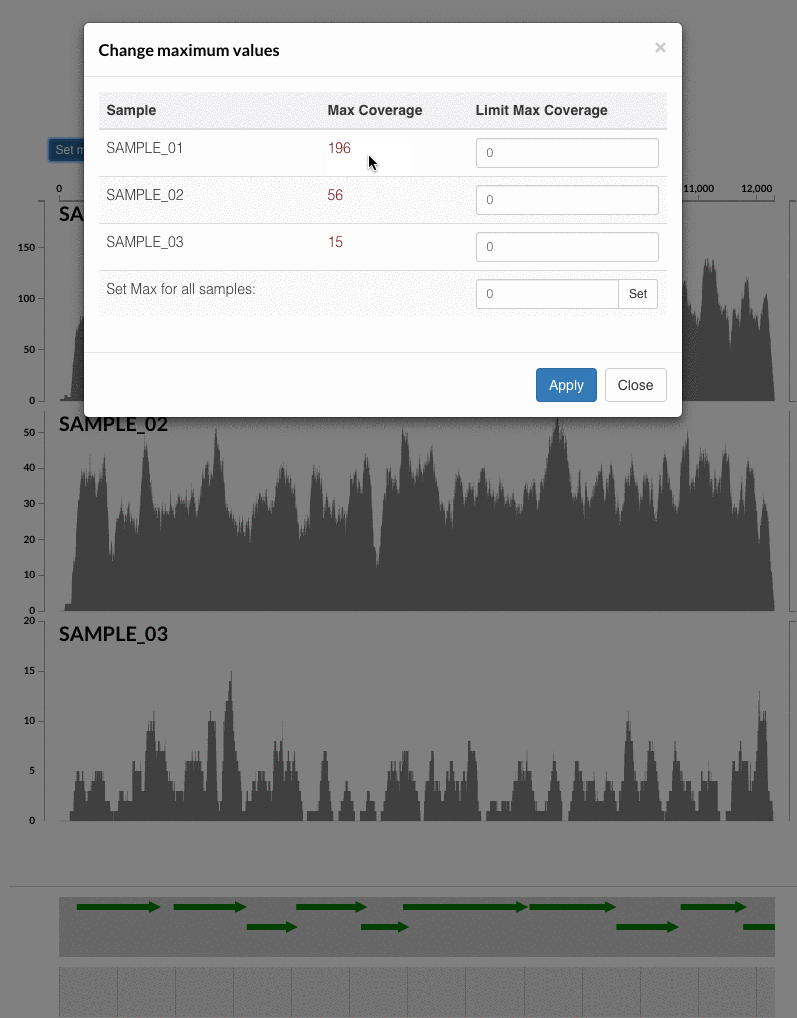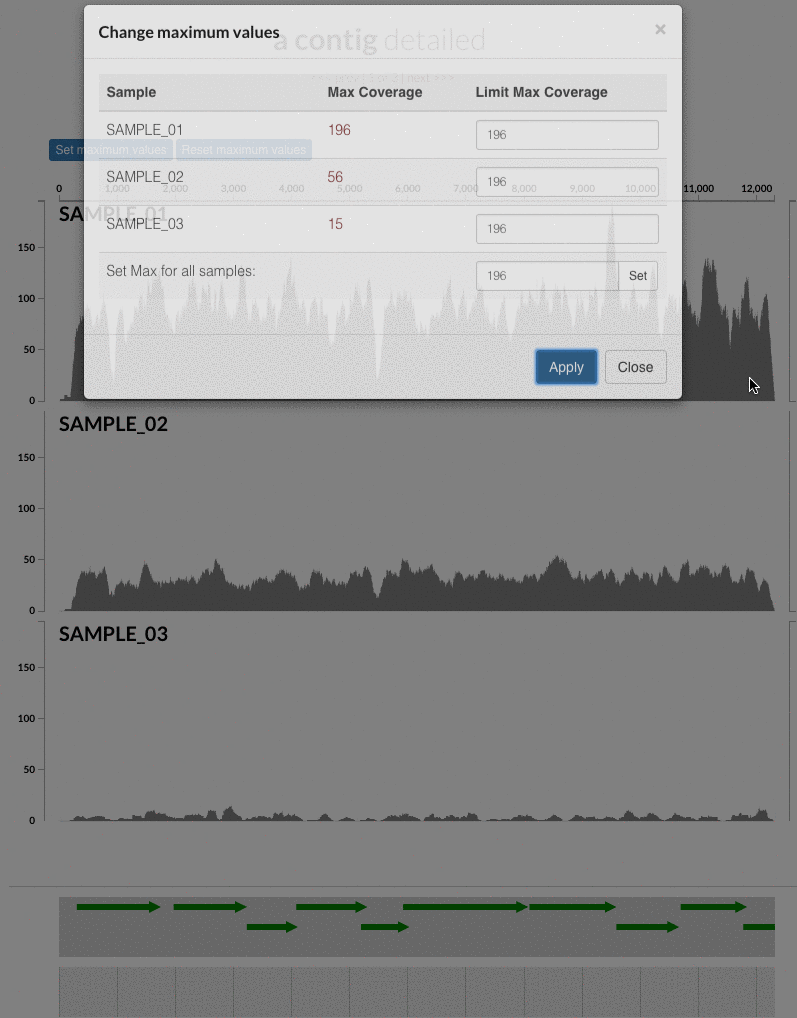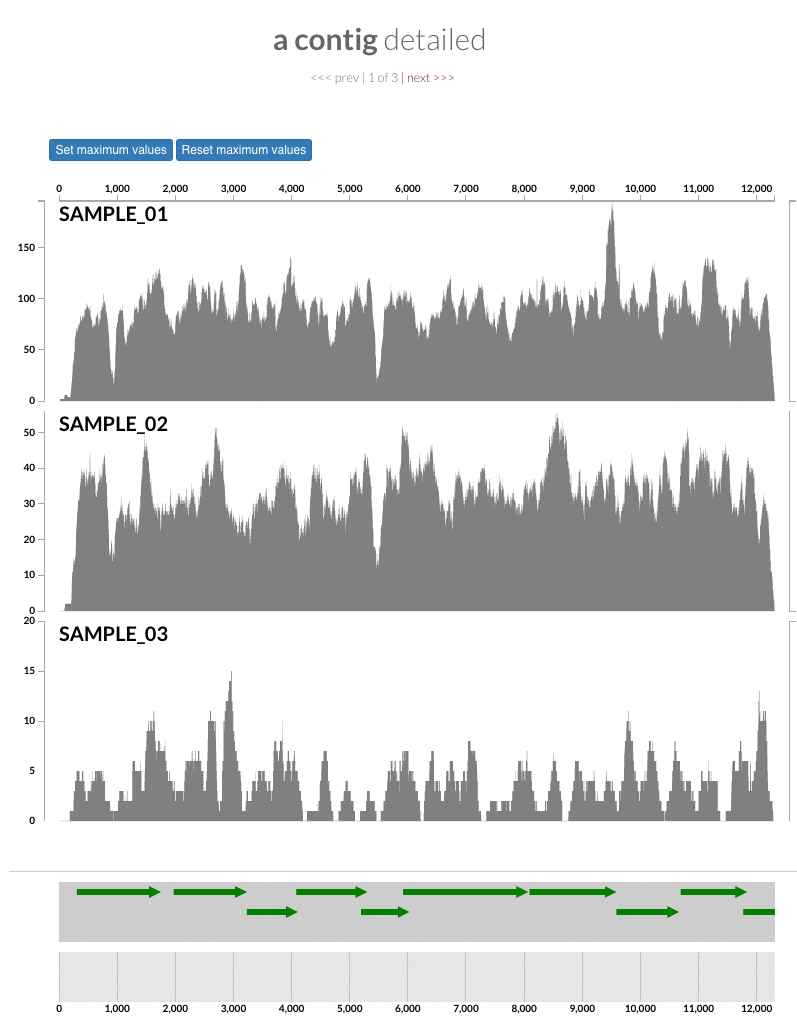
Descriptions tab gets 1-up. One of the most useful features of the interactive interface is the "Descriptions" tab. Yes, we know you are not using it, but you should. Here is an example to see why you should use them (just wait until the page loads, and see all the information that will show up in the right panel): https://anvi-server.org/merenlab/dwh_o_desum. The description tab is extremely useful to take notes and store them in a profile database to remember later. With this new version, you will be able to point out to an item (#715), which will give access to the reader so they can see where it is on the display by highlighting it, or they can inspect it by clicking 'inspect':

Gene mode: a new, highly-resolved interactive mode to study genome bins
This is yet another way for you to examine your data in high-resolution.
We added a flag to anvi-interactive: --gene-mode. When you use this flag along with a collection and a bin name, it allows you to load the interactive interface in the "gene mode". In this mode every item is a gene, instead of a contig, and you can see the coverage, detection, non-outlier coverage, and non-outlier standard deviation of coverage statistics per gene, independently. You can use these data to order the genes, and order the samples. Inspection of nucleotide level coverages, gene sequences, and even gene functions could also be explored in this mode. This allowed us to easily recognize genes that recruit a lot of non-specific mapping, and identify hyper-variable regions in our genomes. One can also search for genes with certain functions, and see their coverages, and the coverage of the genes that are next to them.
Please refer to the help menu for the interactive interface (via anvi-interactive -h or here) to find out more about this mode.
We are excited about this new feature, and we plan to expand it in future versions of anvi'o. If you have any suggestions/complaints/compliments please leave a comment in this issue: #754. We will soon put a tutorial for this mode onli...
anvi'o v3, "Eden"
We are happy to announce a new version of anvi'o, "Eden".
After more than 300 changes that introduced about 6,000 new lines, and removed about 2,250 from the anvi'o codebase, the current version includes many bug fixes, as well as some new features. This release note intends to give you a summary of most important changes.

The codename, which was suggested by @watsonar and won the popular vote, is to mark the arrival of the newest honorary member of our lab. Despite her very young age which doesn't even round to a positive integer, she managed to tip the scale of the gender diversity in our lab for the better. We thank Alon and Rebecca for allowing us to witness essential beauties of life through their happiness.
A new program: anvi-display-contigs-stats
Thanks to @ozcan's recent efforts, anvi'o now can give you basic stats about your contigs databases. Using anvi-display-contigs-stats you can generate insights from one or more contigs databases. Beyond obvious uses, we hope it to be useful to interactively compare different assemblers on the same dataset, or multiple genomes for each of which you have a contigs database.
Since a basic framework is now in place for such comparisons, we will be looking forward to hearing suggestions from anvi'o users to improve it further at every chance.
Here is what you see when you run it on the contigs database of our FMT study:
anvi-display-contigs-stats FMT-CONTIGS.db
And this is what you see when you run it on a bunch of single genomes:
anvi-display-contigs-stats c0328-Microgenomates.db \
c0319-Microgenomates.db \
c0091-Candidate_CPR3.db \
c0205-Parcubacteria.db \
c0661-Parcubacteria.db \
c0792-Candidate_WS6.db
We hope you try it on your current contigs databases, and let us know about your suggestions.
A new versioning approach
We will no longer follow the standards of semantic versioning with anvi'o releases. The last anvi'o version was v2.4.0, this one v3, and the next one will likely be v4, unless there is an absolutely minor change that will not require you to update your v3 installation.
Why leaving the field standards? Anvi'o is an essential tool for us to do science, and we know of multiple other groups besides our's that also use this platform quite rigorously to go after their own questions. Since this is not a media player, which would continue to play your favorite shows even if you don't keep it up-to-date, and since the excellence of everything we do with anvi'o depends on its accuracy, we want our users to update their installations every time there is a new version. When this is the case, the conventional versioning of software becomes rather irrelevant, and somewhat confusing. We are aware of the fact that installing and updating software could be quite a frustrating task, and we do our best to improve those steps for you. If you have any questions or suggestions, please send them to the anvi'o discussion group.
Making a release is quite a painful process, and I personally hate it passionately as it takes at least a full day from my life. We are only doing it when there is a need for it. So, please keep your anvi'o up-to-date while @ozcan, myself, and other developers and contributors do their best to try to keep it bug free for you :)
Bug fixes, new anvi'o programs, and new flags
This version includes a large amount of bug fixes and minor improvement of available programs with additional flags.
New programs that became available with this release include anvi-delete-state, anvi-display-contigs-stats, and anvi-export-samples-db.
We also added new scripts to anvi'o distribution, such as anvi-script-add-default-collection (to add a quick 'default' collection to access to all splits in a profile database, and anvi-script-filter-fasta-by-blast (to remove weak hits from BLAST search outputs).
Some of the new flags we added to our existing programs include --return-codon-frequencies-instead to be able to get codon frequencies instead of amino acid frequencies from BAM files via anvi-get-aa-frequencies, --items-order to explicitly provide an items order to anvi-interactive, --max-num-genes-missing-from-bin and —min-num-bins-gene-occurs to remarkably improve the functionality of anvi-get-seqeunces-for-hmm-hits to filter weak genomes or genes for better phylogenomic analyses thanks to Ryan Bartelme's suggestions, and --align-with flag so we can optionally use FAMSA instead of muscle, which works exceptionally better than muscle in our experience to align sequences within our protein clusters. We heard FAMSA thanks to Antonio Fernandez-Guerra.
Contigs database version bump
We realized that were. You will be able to update your existing contigs databases without any pain via the upgrade script anvi-script-upgrade-contigs-db-v8-to-v9.
Genome storage version bump
We extended the functionality of anvi'o genome storages that are used for pangenomic workflows. They now also keep a copy of every gene they describe in DNA alphabet. Unfortunately this also requires an update, which can be done via anvi-script-upgrade-genomes-storage-v3-to-v4.
Thank you for your interest in anvi'o!
Please find anvi'o tutorials and installation instructions here:
anvi'o v2.4.0, "Pyrenees"
We are happy to announce the new version of anvi'o, "Pyrenees".
After 350 changes in the codebase that introduced more than 4,500 lines of code and removed about 9,000, the current version includes many bug fixes, as well as some important additions to the repository. This release note will give you a summary of most important changes.

The codename is to honor our friend and colleague, Tom Delmont, who is going to continue working with us from France. The Pyrenees is a mountain range in southwest Europe between France and Spain, where Tom spent most of his life.
Single-amino acid variants (SAAVs)
Since its very first version, anvi'o has been providing you with one of the most comprehensive frameworks to make sense of single-nucleotide variants (SNVs). Here is a tutorial for skeptics.
Although it has been in anvi'o for more than a year, we are very excited to officially announce the anvi'o workflow to investigate single-amino acid variants. More resources on SAAVs will soon be available, and we will keep you posted. But if you have been using the anvi-gen-variability-profile program to characterize single-nucleotide populations in your population genomes, all you need to do now is to add --engine AA to get single amino acid profiles.
Fluency in phylogenomics
Anvi'o now can speak phylogenomics. Here you will find an extensive tutorial with reproducible examples:
http://merenlab.org/2017/06/07/phylogenomics/
You now can do phylogenomic analyses in anvi'o for metagenomic bins or pangenomes thanks to others who pushed us for it. Luke McKay of the Montana State University asked for gene concatenation for HMM hits in genomes from metagenomes, we delivered it, (47fd334, b9780a5, 29a3827, 3ee670e, fa7128a). Then, Ryan Bartelme of the University of Wisconsin-Milwaukee asked for concatenated genes in protein clusters of pangenomes by sending a private e-mail, and we delivered it, too (1726476, 3d07e8b, 99359bc). We didn't stop there. We implemented a simple driver for FastTree (227e892) for starters so you can immediately start playing with your data. As always, we are thankful for your suggestions.
Identifying ribosomal RNAs everywhere
In most cases, getting ribosomal RNA genes out of isolate or metagenome-assembled genomes is not as straightforward as one would like, even when the assembler managed to assemble them.
Gene callers usually don’t perform well when it comes to identifying 16S or 23S rRNA genes, and using primer sequences for these regions is not exactly 2017. We added a new feature in anvi’o that reduces the recovery of rRNA gene sequences from isolate, single-cell, or metagenome-assembled genomes to a couple of key strokes. More information and examples are here:
http://merenlab.org/2017/07/09/recovering-rRNAs/
The small but mighty 'push' button
This is something we are very excited about. You know how sometimes you have something on your interactive interface you would like to show to your colleagues, or share with everyone on the planet? Well, with this version you will be able to click on this little new cloud button on the interface,
and you will be able to send your interactive display directly to http://anvi-server.org or any other anvi'server instance online. After which you can share this interactive display in read-only mode privately with your colleagues, editors, or reviewers, or with everyone by making it public.
Here is an appropriate use of it in a recent paper in Cell Reports that gives another dimension to a static figure:
https://www.youtube.com/watch?v=WtPB9GchuEI
More functional side buttons
Now we have a way to deliver some important news to your door step:
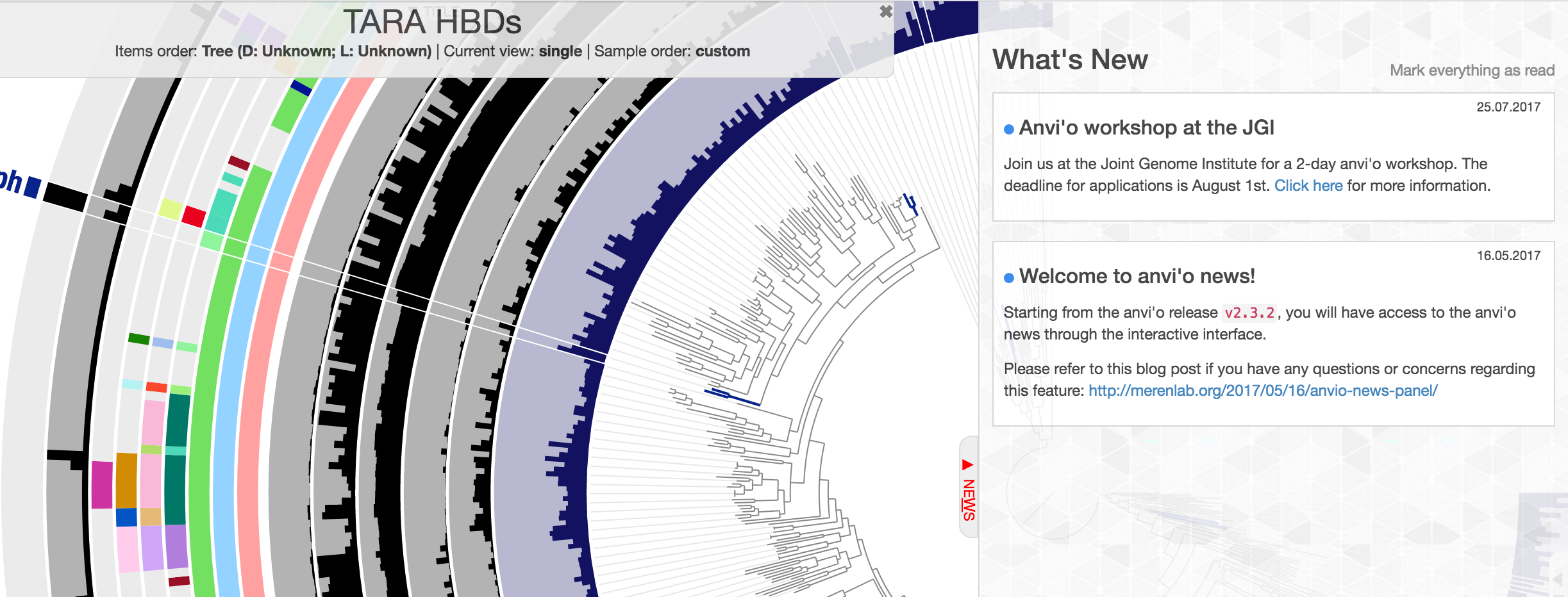
More information on this new addition, including notes about possible privacy concerns is here:
http://merenlab.org/2017/05/16/anvio-news-panel/
Please consider sharing your thoughts and opinions on this.
We also made sure anvi'o can convey extensive descriptions of the displays shown in anvi'o. Here is an example:

The description tab gives access to an editor, in which you can describe the data using Markdown syntax. We hope that this practice catches on, so whenever someone looks at an anvi'o display, they know that there will be some information on the side panel to better understand the details.
Please find anvi'o tutorials and installation instructions here: