anvi'o v5, "margaret"
We are happy to announce a new version of anvi'o, "margaret".
After nearly 1,500 changes that introduced about 15,000 new lines to the anvi'o codebase and removed about 4,000 from it, the current version includes many fixes to big and small bugs, as well as new features. This page intends to give you a summary of most notable changes that comes with margaret.

The codename is a small tribute to Margaret Oakley Dayhoff, an American physical chemist, who is known as the founder of bioinformatics. Dayhoff developed first programmable computer methods to compare protein sequences, and published in 1965 a book titled "Atlas of Protein Sequences and Structure", which is considered as of today the first text book of bioinformatics. The codename was suggested by Mick Watson, and won the popular vote on Twitter. Dayhoff sadly died at an early age of 57 in 1994, shortly before bioinformatcis emerged as a distinct field. However, her astonishing contributions to life sciences, such as the development of essential approaches for protein sequence comparison and evolutionary tree construction, still constitute some of the most common approaches in our bioinformatics toolkit.
Your new disconcerting toy: GC-content overlaid on reference contexts
Metagenomic read recruitment often results in wavy coverage patterns in the reference context. This phenomenon, which can be attributed to three major sources, can result in up to an order of magnitude coverage difference for genes within the same contig. While we are kind enough to leave those alone who solely work with metagenomic short reads to quantify functions in metagenomes in their blissful world, we wanted to include in this version of anvi'o something so you can overlay GC-content change throughout your contigs to see whether variation you observe in the context of some of your key genes is largely driven by GC-content or not:

This is not yet anything but a qualitative insight for you to make sense of to what extent variation in coverage could be explained by deterministic factors that have nothing to do with the biology of your system given the metagenome, but it shows that more quantitative insights into this could be useful. We will think about this going forward, and we are open to your suggestions!
A new anvi'o workflow management system for serious anvians
This new version of anvi'o includes a new program anvi-run-workflow, which provides an interface to our new module that implements snakemake-based anvi'o workflows.
These workflows offer accessible, reproducible, and comprehensible solutions for complex analyses that may include hundreds of samples. We have been using anvi-run-workflow every day in our lab since it first appeared in our master repository, and we are happy to make its power available to you as soon as we could.

There will be an extensive tutorial very soon, but until then you can send your questions to Alon (smiley).
Single-codon variants for a more powerful framework to study microbial population genetics
Anvi'o already could make sense of single-amino acid variants (SAAVs) in environmental metagenomes. But working with SAAVs was limiting our ability to infer and quantify neutral processes that may not result in changes in the amino acid sequence. We changed our design in such a way, now anvi-profile can characterize single-codon variants (SCVs) if --profile-SCVs flag is declared. We updated our reference manual for variability analysis to include new sections describing SCVs and SAAVs.
With SNVs, SCVs, and SAAVs, anvi'o v5, deserving of its codename, offers a robust framework to investigate population genetics of environmental microbes, while SCVs and SAAVs leverage our ability to tease apart evolutionary forces acting upon them. We hope you enjoy these new toys, and feel free to get in touch with us if you have questions or suggestions.
Visualize environmental variation on protein structures through the new Structure DB
Our efforts to push the boundaries of investigations of environmental variation within microbial populations reaches to a new level in this release with a brand new ability about which we are very excited: linking variation to predicted protein structures.
With the new structure database associated workflows anvi’o can predict the tertiary structure of genes identified from a contigs database using the Protein Database Bank. Then, it can directly overlay onto the predicted protein structures the variability data from your metagenomes in the form of SCVs and SAAVs. All of this is accomplished in just two new programs, anvi-gen-structure-database and anvi-display-structure.
We believe that this nexus between structural biology and metagenomics will elevate environmental metagenomics into the realm of biophysics, and enable investigations into evolutionary processes driving the diversity of proteins that could not be learned from sequence analyses alone.
With these new advances come two new dependencies to additional open-source software, for which we are very grateful: MODELLER and DSSP.
Here is a teaser from the new interactive anvi-display-structure interface:

We will soon make available an extensive tutorial to describe this workflow in detail. Until the, you can send your questions to Evan and Ozcan.
Computing average nucleotide identity for genomes in pangenomes
This release also includes significant improvements for our comparative genomics and pangenomics workflows.
One of these improvements is the inclusion of a new program, anvi-compute-ani, to calculate the average nucleotide identities across a given set of genomes, which can be automatically added into any anvi'o pangenome.
For instance, this is an anvi'o pangenome of the 31 Prochlorococcus isolates we played with in our recent paper:
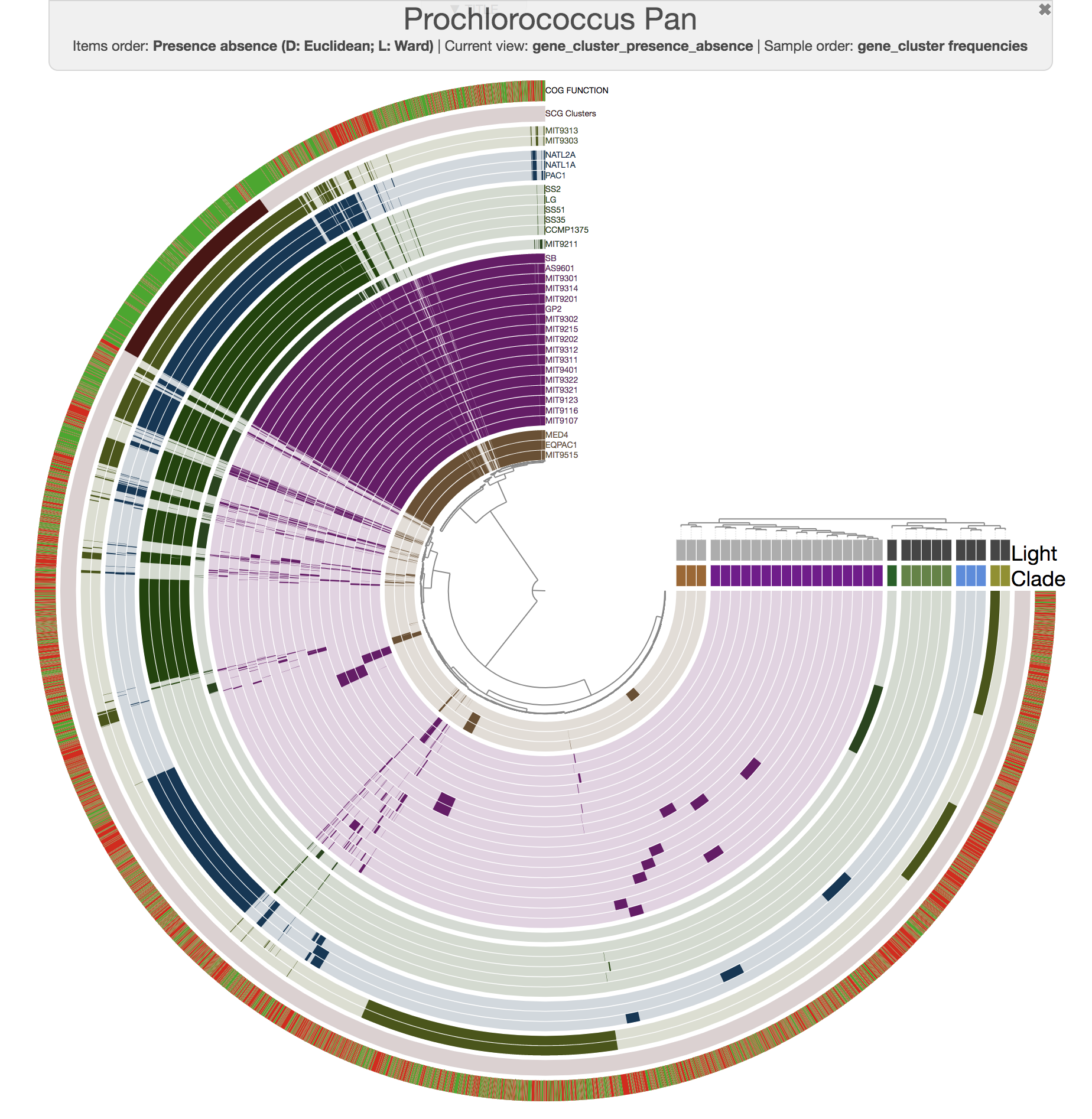
And this is what you get when you run anvi-compute-ani:
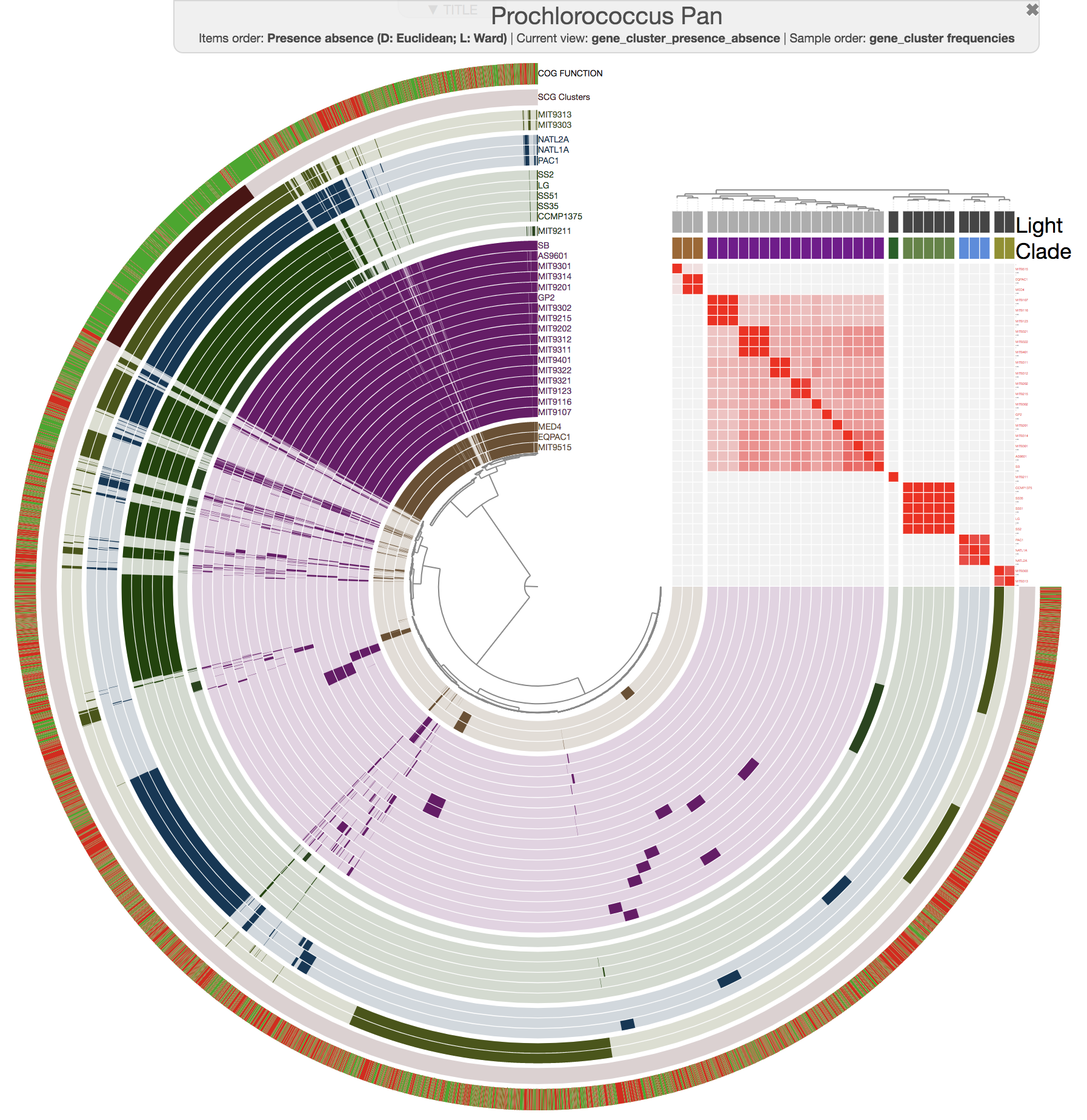
Mike Lee had suggested this as an option a long time ago. We are happy to finally deliver this functionality, which uses pyANI as a backend, for which we are thankful for its developers.
We updated our tutorial on pangenomics to describe intermediate steps.
A new approach to explore functional enrichment in pangenomes
This version of anvi'o also incluedes a new analytical framework to study functional enrichment in a given pangenome based on any arbitrary organizations of genomes. You simply define how would you like to partition your genomes, whether based on a phylogenetic tree or a dendrogram that anvi'o computed from gene cluster distributions, and this new tool finds functions that are enriched in those groups (i.e. functions that are characteristic of a given group of genomes, and predominantly absent from genomes from outside this group).
This is done by the new program anvi-get-enriched-functions-per-pan-group, and Alon extended our current tutorial on pangenomics with an extensive description of how it works.
Native functional annotation options += PFAMs
If you have your own functional annotations for your genes in an anvi'o contigs database, it is quite straightforward to import them via anvi-import-functions program. Anvi'o v3 had made available another program to automatize the annotation process, anvi-run-ncbi-cogs, if you were fine with NCBI's Cluster of Orthologus Groups. This release contains a new program, anvi-run-pfams to use the collection of HMMs produced by the European Bioinformatics Institute based on UniProt.
Tree modification through the interactive interface
It has been a challenge to deal with phylogenetic tree operations in anvi'o interactive interface. This version includes a significant code refactoring effort, which makes possible to have new toys that we could not have before. These new toys include basic tree editing and storage abilities such as re-rooting trees, rotating and collapsing branches. You can even see the branch support values in the mouse tab of the anvi'o interactive interface. These functions are now available to you through the menu that appears when you click a branch in the interactive interface while pressing the Command or Control key:

A new HMM collection to estimate completion of eukaryotic bins
Since its conception anvi’o included single-copy core gene collections to assess the completion and redundancy of bacterial and archaeal bins. This release includes a collection to estimate the completion of eukaryotic bins that Tom Delmont, who recently left us physically to join the ranks of Genoscope, curated from the BUSCO collection.
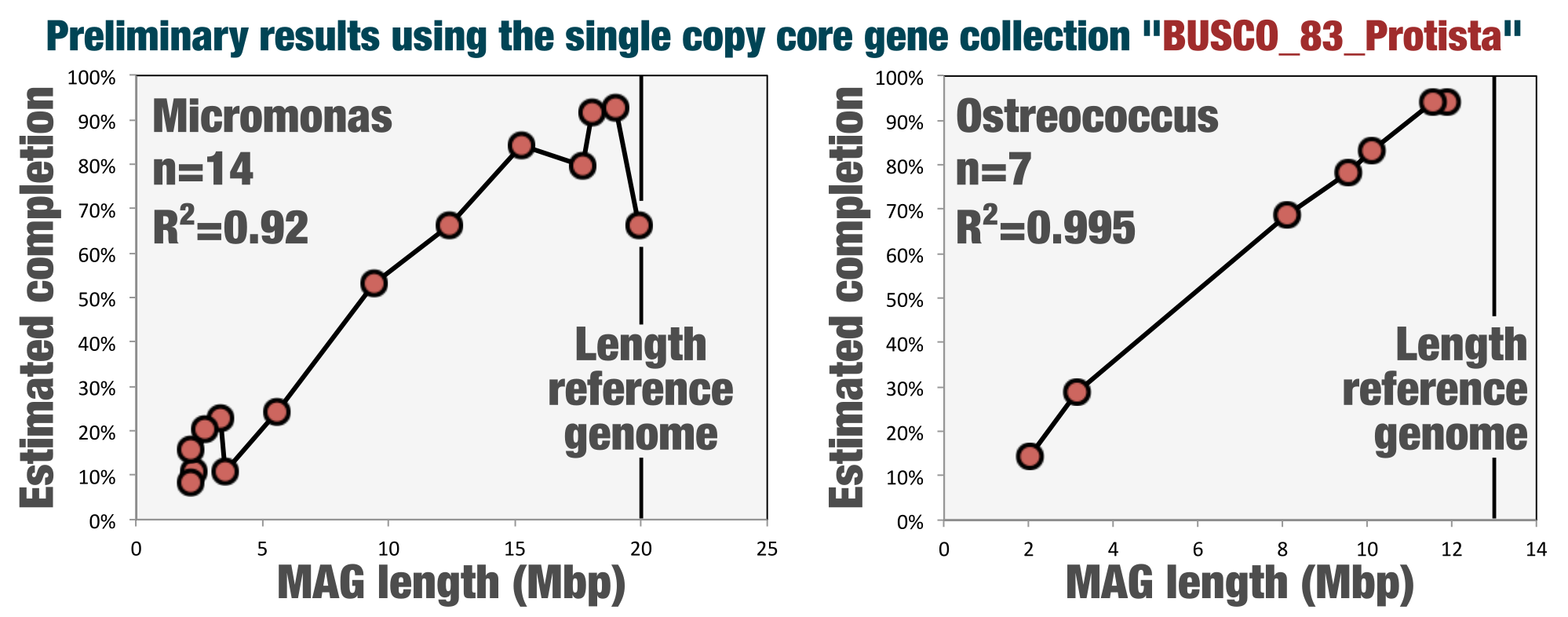
See Tom's blog post for details and preliminary benchmarks (also, if you are finding these release notes too boring to read, you can try reading this one too).
If you are recovering tiny eukaryotic organisms from your metagenomes please help us improve this collection by reporting back your experiences with it.
Importing metagenome-level short-read taxonomy and the enhanced stacked bar data type
While our efforts on shotgun metagenomes largely focus on genome-resolved strategies, we acknowledge that one could learn a lot from taxonomic annotation of short-reads as an additional layer of information. In this release anvi'o comes with a new program, anvi-import-taxonomy-for-layers with a KrakenHLL parser, which can import short-read level taxonomic annotations into anvi'o profiles. Thanks to the improved data groups, different levels of taxonomy would be available in the layers tab,

And could be visualized easily:

The best part is that our improved stacked bar data type in this release then would allow you to order your metagenomes based on the relative abundance of any given taxon at any given taxonomic level in those metagenomes according to short reads (the example below, orders metagenomes in the infant gut dataset from Sharon et al. based on the increasing relative abundance of Enterococcus):
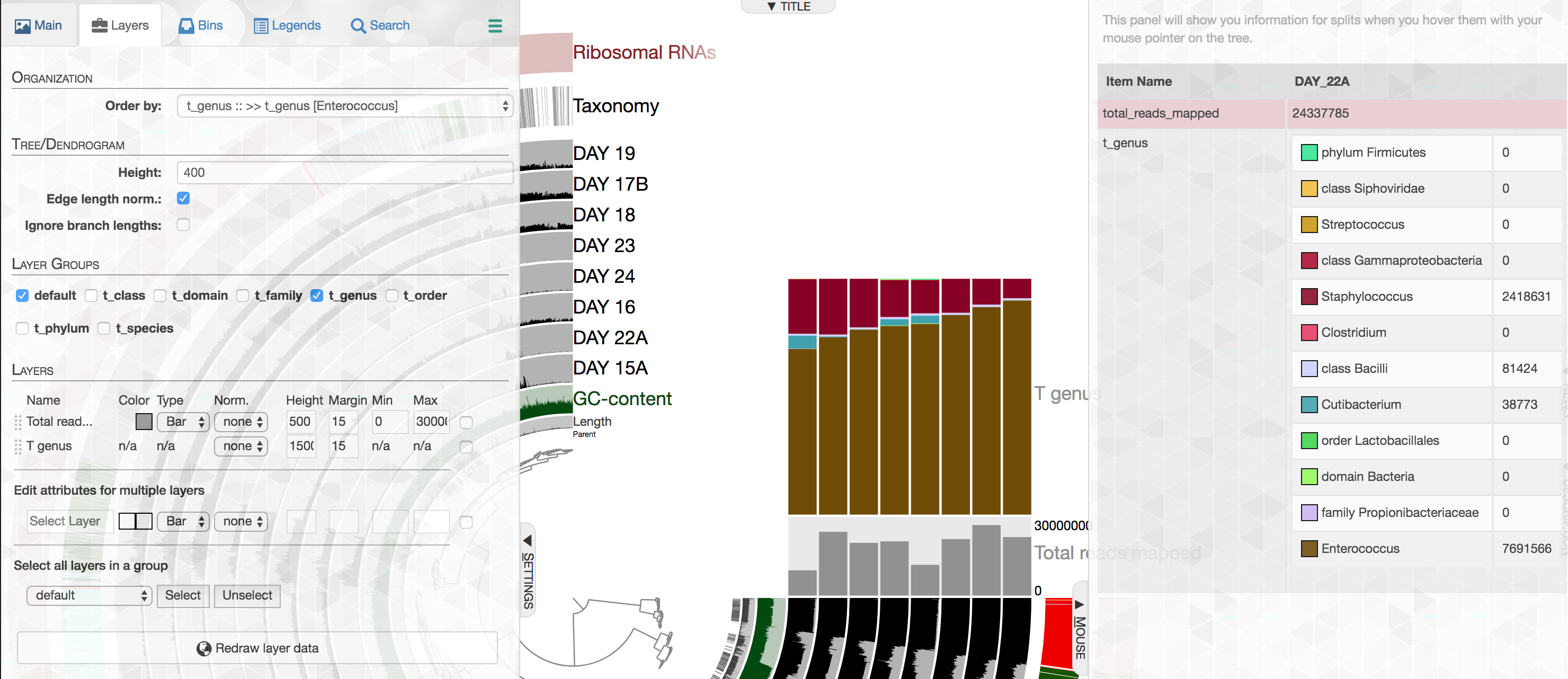
Here we would like to assume that you're saying to yourself "the example is boring, but the concept has promise". Thanks! We agree.
Thanks
A year ago I listened to Jeff Gordon's talk at the University of Chicago to which he started with this African proverb:
If you want to go fast, go alone. If you want to go far, go together.
This concept applies to scientific endeavors so well. Speed is transient, and teamwork is essential for major contributions. Fortunately anvi'o has been becoming more and more of a team effort. But looking at our release notes, I don't know whether we could go any faster from v4 to v5 either. This release was a result of significant intellectual and coding contributions from Alon Shaiber, Evan Kiefl, as well as Özcan Esen, whose guidance and hard work continue to keep this operation together. Altogether, they spent hours and hours on big and small features and issues, with an enthusiasm that can be best justified by curiosity and the desire to contribute to your journey in data-driven microbiology. I, Meren, who gets to write this release note one more time, thank them wholeheartedly.

As a team we also thank Jarrod Scott, Alexandra Campbell, Samantha Atkinson, Carlos Ruiz, Bryan Merill, Mike Lee, Varun Srinivasan, and many others who asked for features and reported bugs with their endless patience with us.
We hope you find v5 useful for your research, and we certainly hope you will not run into any bugs we probably left in the code 😇
If you are interested in anvi'o but don't know where to start, catch us in one of our free workshops, or find us on our Slack channel.