An experimental project for mapping genomic data onto 3D protein structures in Jupyter Notebooks.
The Jupyter Notebooks in this repository can be run in your web browser using two freely available servers: Binder and CyVerse/VICE. Click on the buttons below to launch Jupyter Lab. It may take several minutes for Jupyter Lab to launch.
Binder is a platform for reproducible research developed by Project Jupyter. Learn more about Binder. There are specific links for each notebook below, however, once Jupyter Lab is launched, navigate to any of the other notebooks using the Jupyter Lab file panel.
Binder provides an easy to use demo environment. Due to limited resoures, Binder is not suitable for compute or memory intensive production analyses and may occasionally fail to run the notebooks in this repository.
NOTE: Authentication is now required to launch binder! Sign into GitHub from your browser, then click on the launch binder badge below to launch Jupyter Lab.
The new VICE (Visual Interactive Computing Environment) in the CyVerse Discovery Environment enables users to run Jupyter Lab in a production environment. To use VICE, sign up for a free CyVerse account.
The VICE environment supports large-scale analyses. Users can upload and download files, and save and share results of their analyses in their user accounts (up to 100GB of data).
![]()
Follow these step to run Jupyter Lab on VICE
The notebooks in sars-cov-2 folder map missense mutations aggregated by the COVID-19-Net Knowledge Graph to available 3D protein structures in the Protein Data Bank. Mutations are mapped onto protein-protein interaction sites, ligand binding sites, drug binding sites.
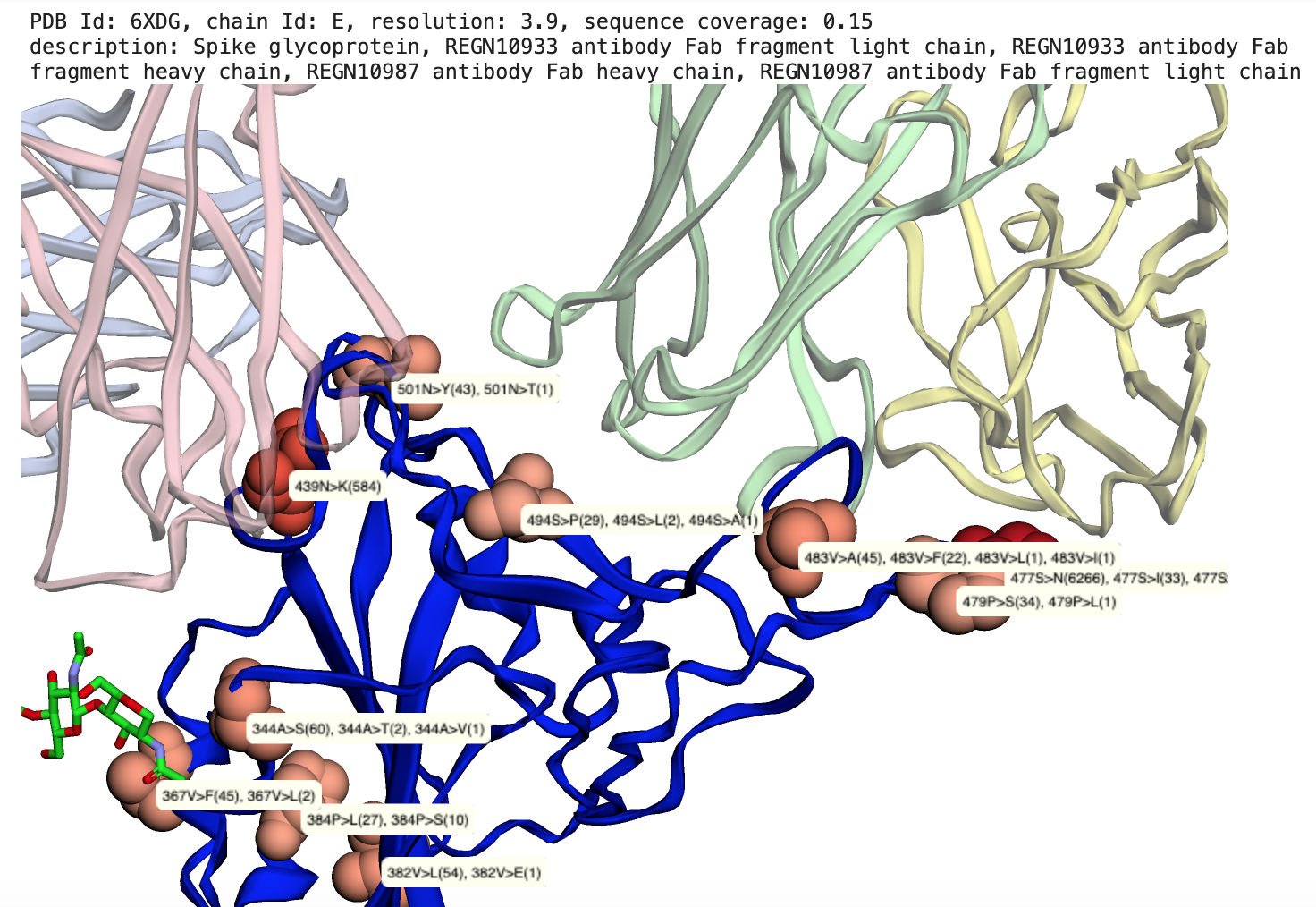 |
Map SARS-CoV2 mutations to 3D structures, Example: Two Regeneron Fab fragments bound to Spike glycoprotein RBD (REGN10933-RBD-REGN10987 complex (1)) with observed mutations highlighted |
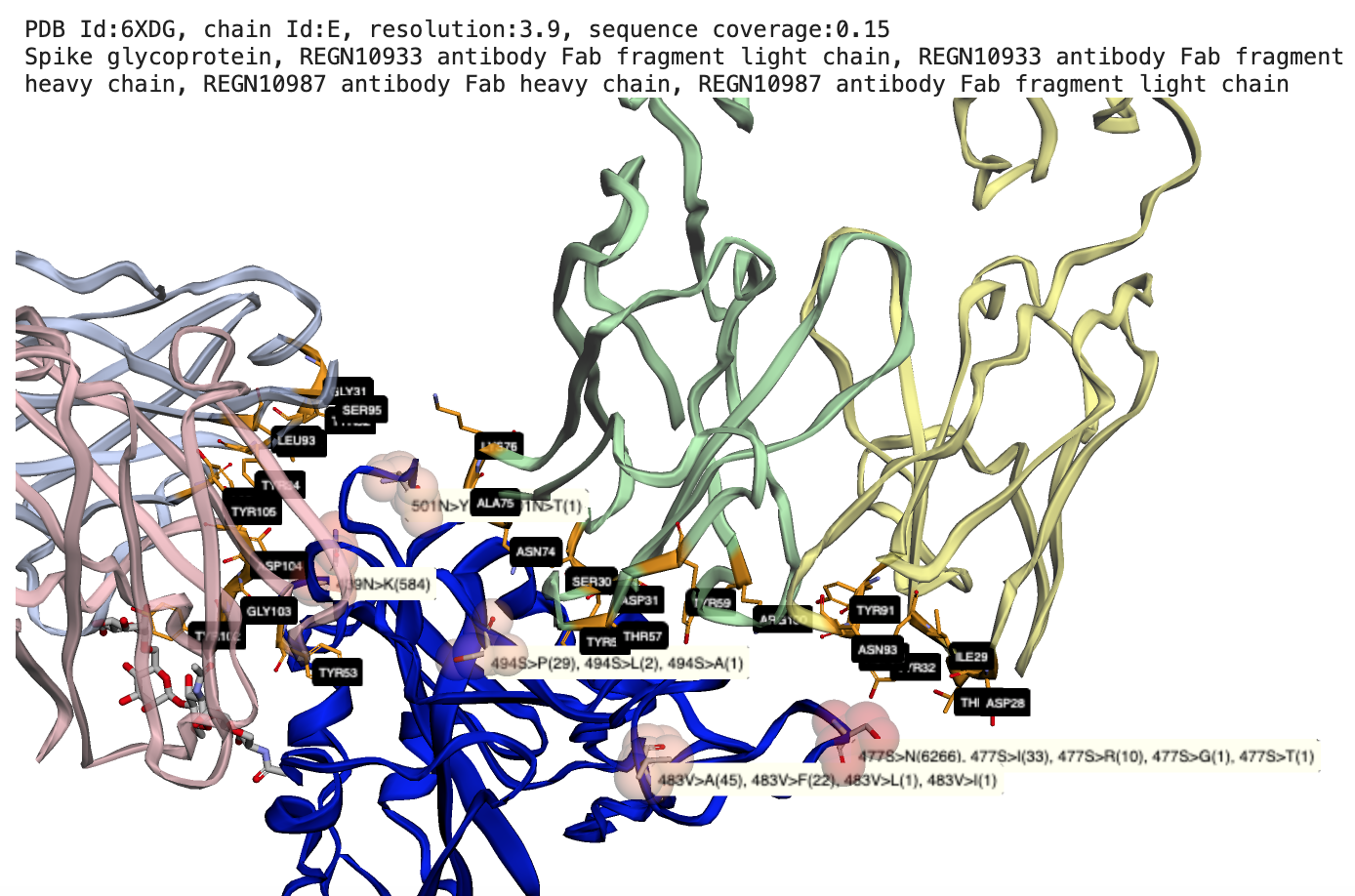 |
Map SARS-CoV2 mutations to protein-protein interactions, Example: Two Regeneron Fab fragments bound to Spike glycoprotein RBD with observed mutations at the binding interface |
Reference: (1) Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010-1014. doi:10.1126/science.abd0827, PDB id: 6XD6.
The notebooks below visualize the positions of missense mutations mapped from dbSNP to 3D protein structures in the Protein Data Bank. Variations can be filtered by the clinical significance level from ClinVar, UniProt Ids, or a list of specific variants specified by the rs identifier or genomic location.
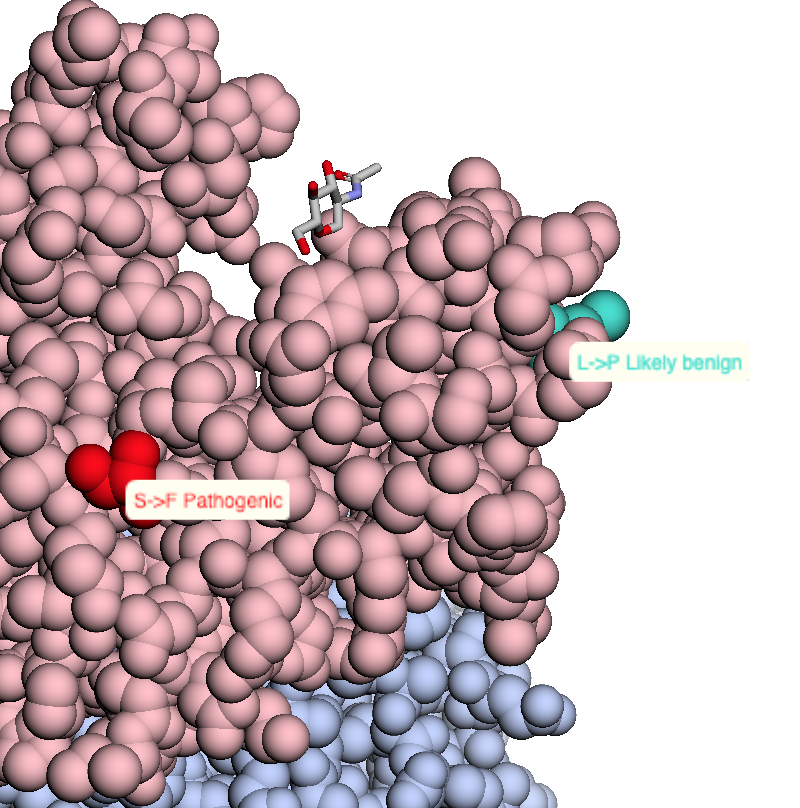 |
Map missense mutations from dbSNP to 3D structures |
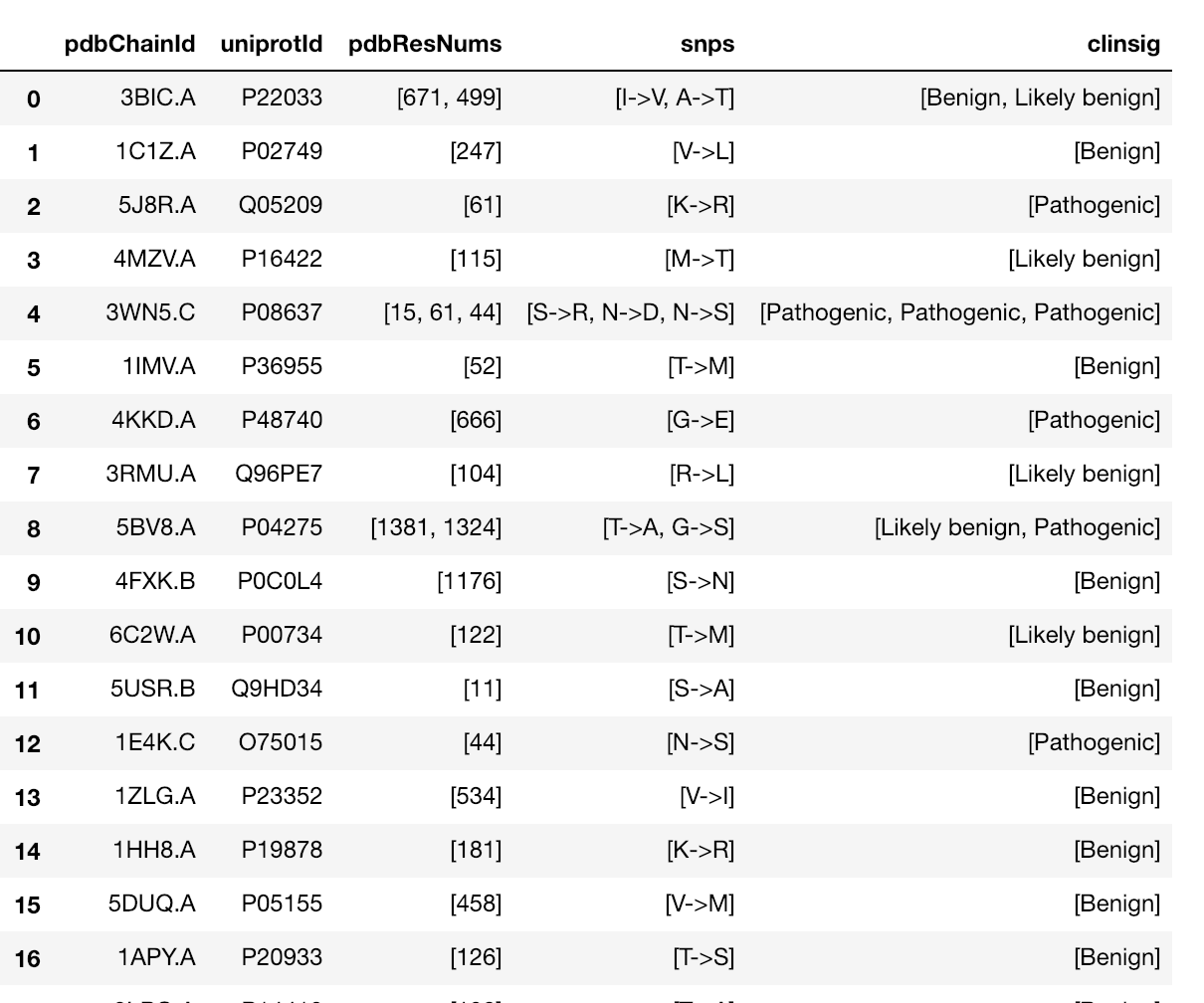 |
Map missense mutations from dbSNP to 3D structures that contain the associated amino acid change |
This notebook maps a dataset of 63,197 missense mutations with allele frequencies >=1% and <25% extracted from the ExAC database to 3D structures in the Protein Data Bank. The dataset is described in:
Niroula A, Vihinen M (2019) How good are pathogenicity predictors in detecting benign variants? PLoS Comput Biol 15(2): e1006481. doi: 10.1371/journal.pcbi.1006481
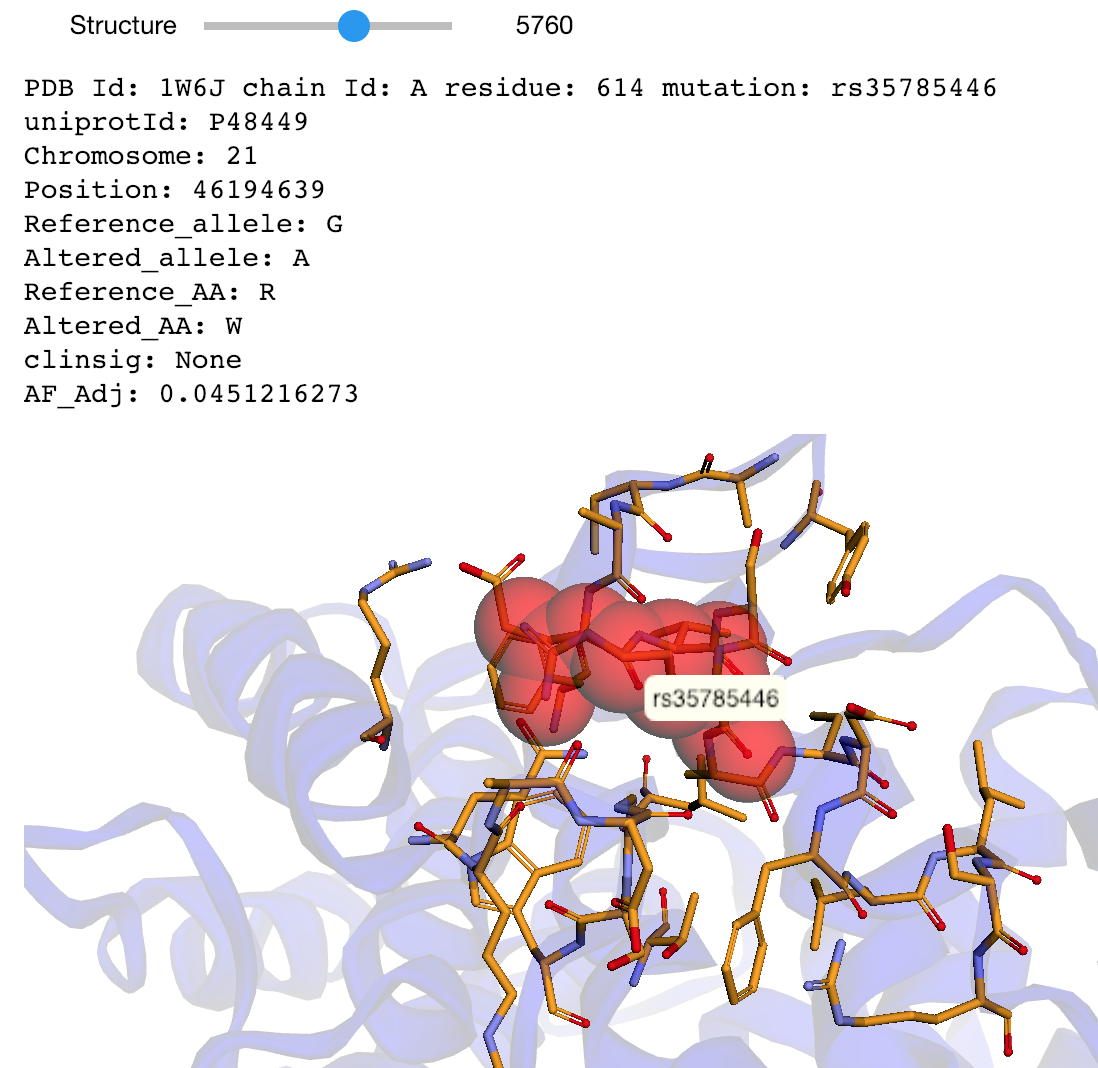 |
Map mutations with high allele frequences to 3D structures |
This protype pipeline demonstrates how to map genetic locations of SNVs to 3D structures. To run this demo, click on the "launch binder" link below. At the bottom of each notebook is a link to the next step. In total, there are 5 steps to this pipeline, shown below.
By replacing the demo input file with your own data and adjusting the notebook that reads the data, you can run our own custom analysis.
 |
Read and standardize genetic location data |
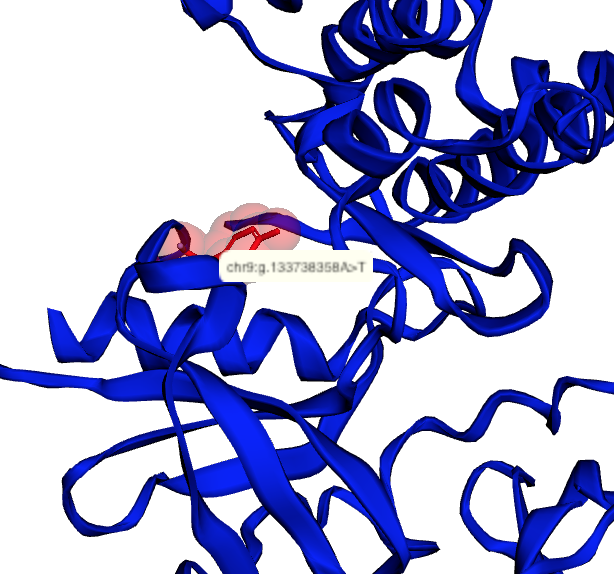 |
Map genetic locations to 3D protein structures |
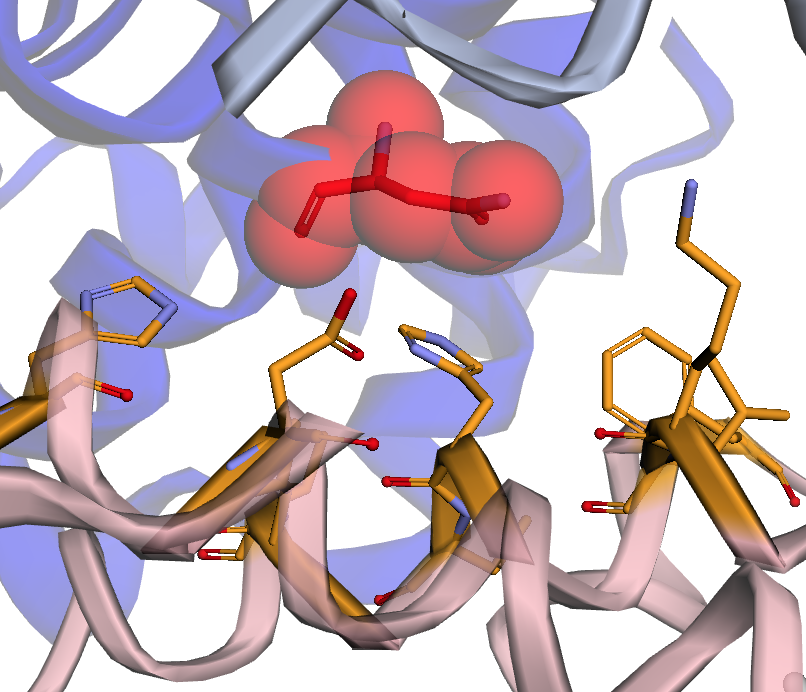 |
Map genetic locations to protein-protein and protein-nucleic acid interfaces |
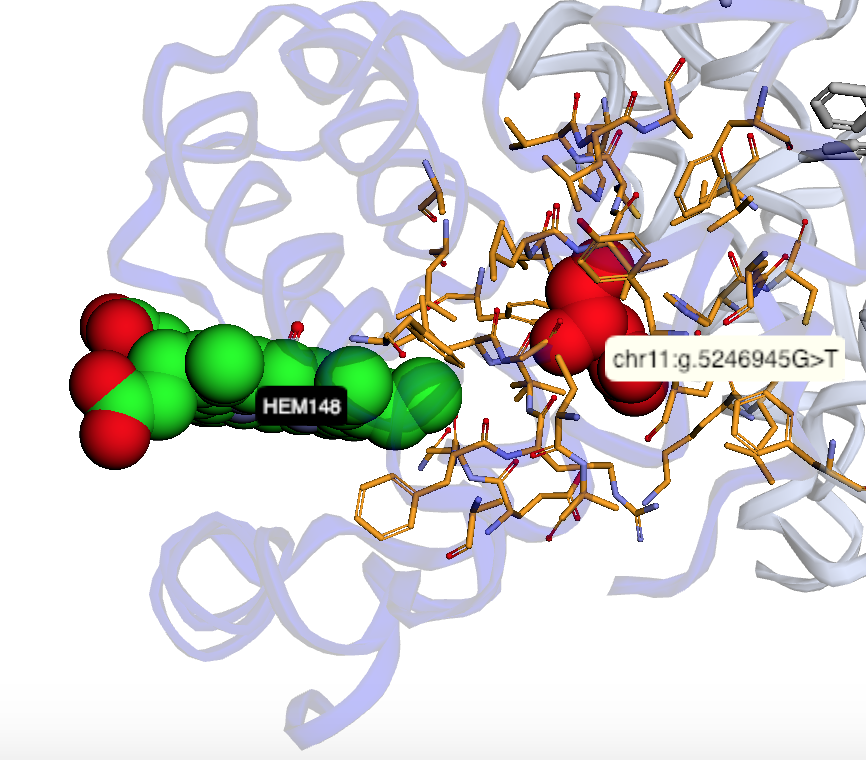 |
Map genetic locations to ligand binding sites |
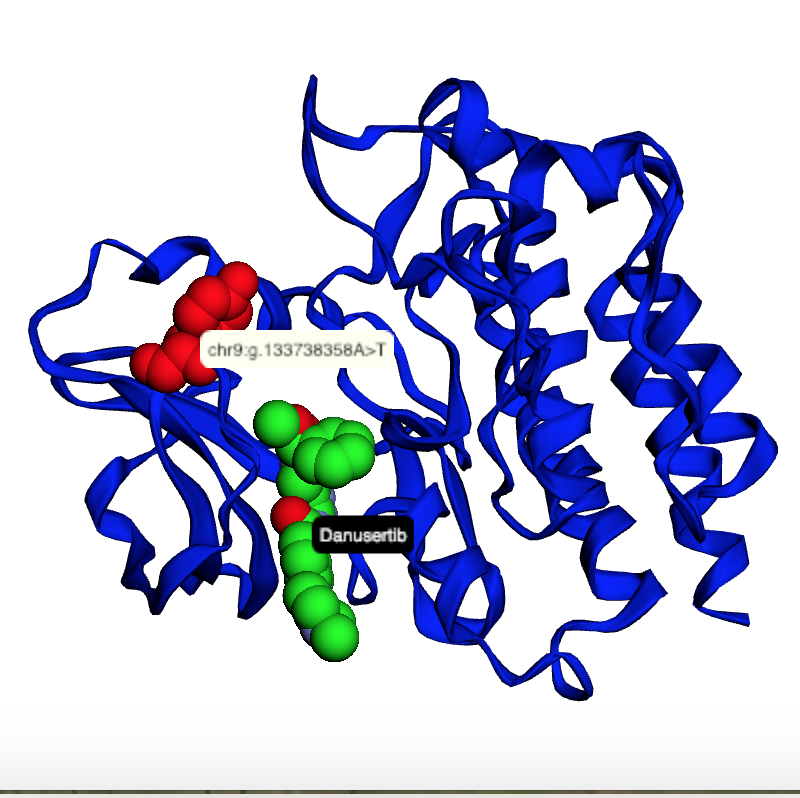 |
Map genetic locations to drug binding sites |
Please send feedback or feature requests.
Interested in a collaboration? Please send us use cases.
Bhattacharya R, Rose PW, Burley SK, Prlić A (2017) Impact of genetic variation on three dimensional structure and function of proteins. PLoS ONE 12(3): e0171355. doi: 10.1371/journal.pone.0171355
Bradley AR, Rose AS, Pavelka A, Valasatava Y, Duarte JM, Prlić A, Rose PW (2017) MMTF - an efficient file format for the transmission, visualization, and analysis of macromolecular structures. PLOS Computational Biology 13(6): e1005575. doi: 10.1371/journal.pcbi.1005575
Glusman G, et al. (2017) Mapping genetic variations to three-dimensional protein structures to enhance variant interpretation: a proposed framework. Genome Medicine 9 (1), 113. doi: 10.1186/s13073-017-0509-y
Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlić A, Rose PW (2018) NGL viewer: web-based molecular graphics for large complexes, Bioinformatics, bty419. doi: 10.1093/bioinformatics/bty419
Valasatava Y, Bradley AR, Rose AS, Duarte JM, Prlić A, Rose PW (2017) Towards an efficient compression of 3D coordinates of macromolecular structures. PLOS ONE 12(3): e0174846. doi: 10.1371/journal.pone.01748464
Project Jupyter, et al. (2018) Binder 2.0 - Reproducible, Interactive, Sharable Environments for Science at Scale. Proceedings of the 17th Python in Science Conference. 2018. doi: 10.25080/Majora-4af1f417-011
Merchant N, Lyons E, Goff S, Vaughn M, Ware D, Micklos D, et al. (2016) The iPlant Collaborative: Cyberinfrastructure for Enabling Data to Discovery for the Life Sciences. PLoS Biol 14(1): e1002342. doi: 10.1371/journal.pbio.1002342
Goff, Stephen A., et al. (2011) The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Frontiers in Plant Science 2. doi: 10.3389/fpls.2011.00034
Sayers EW, et al. (2019) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 47, D23–D28. doi: 10.1093/nar/gky1069
Wang J, Sheridan R, Onur Sumer S, Schultz N, Xu D, Gao JJ (2018) G2S: A web-service for annotating genomic variants on 3D protein structures, Bioinformatics, 34(11), 1949-1950. doi: 10.1093/bioinformatics/bty047
Rego N, Koes, D (2015) 3Dmol.js: molecular visualization with WebGL, Bioinformatics 31, 1322–1324. doi: 10.1093/bioinformatics/btu829
The MMTF project (Compressive Structural BioInformatics: High Efficiency 3D Structure Compression) is supported by the National Cancer Institute of the National Institutes of Health under Award Number U01CA198942. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The CyVerse project is supported by the National Science Foundation under Award Numbers DBI-0735191, DBI-1265383, and DBI-1743442. URL: www.cyverse.org