An exploration of reinforcement learning with Deep-Q Networks using Sphero robotic balls. Check out Neural.Swarms the learn about training neural network agents. Also, you may be interested in the development discussion from my initial efforts on this project while a data science fellow at Galvanize. If my code or methods seem obtuse, it may explain why. Moving forward, clean code!
- Initial Goals
- Controlling Sphero from Python
- Technical difficulties
- Major utilities
- References
The goals are behavioral in nature, and currently very simple.
- Navigation : using reinforcement learning in a physical space to learn how to get to the center of a webcam image.
- Cooperation : multiple agents locate and move a target to the center of the webcam image.
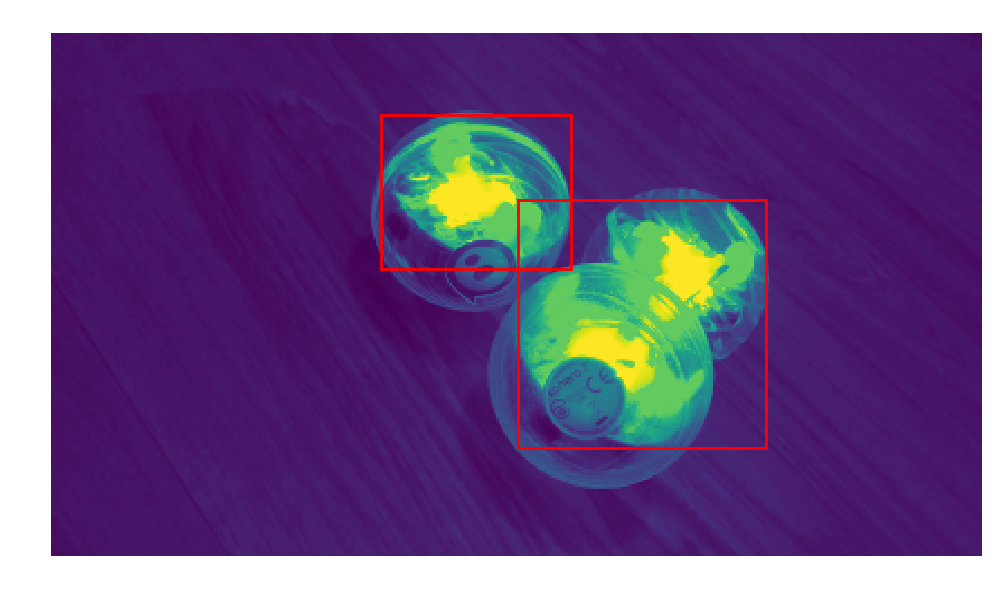
The kulka library provides everything we need to send basic movement commands to Sphero. I added a data polling function to it, so you'll need my fork. This gets us the locator data, but I still haven't gotten full sensor streaming. Thankfully, SKImage includes plenty of easy to use algorithms. In src/webcam_segmentation/py you'll find the methods I used, all standard packages in the SKImage library. With a filter and blob detection, I found the Sphero in the images reliably enough. The filtered image became my neural network inputs for some tests, and in other tests I just used the coordinate as inputs. The current version showing promise uses coordinates from a set of five frames. Once I have good results there, I want to move to a convolutional network on five stacked frames. However, a network that takes coordinates could also take Sphero output data as inputs. Check out the development discussion for more details on the computer vision techniques.
For the simulation models the agent receives rewards on [-1, 1] based on distance from the goal. This helps the agents learn faster than in other contexts, as each state had a distinct reward associated with it. In other domains, like the DeepMind network which plays Atari games, many actions have no rewards associated with them until the agent reaches an end state. This is called the credit assignment problem. If the agent receives rewards only at the end of the game, how can it tell which actions led to that reward? Reinforcement learners solve this through massive amounts of gameplay training, and the Q-function simply learns to implicitly calculate sequences of rewards. In simulation, I can avoid the credit assignment problem entirely with the reward function I've chosen. But, when I deploy that to Sphero, the CV algorithms aren't reliable enough to always give a reward, and sometimes give totally incorrect rewards. Without training an entirely new model just for scoring the RL model, there's no way around this, and the Sphero agent will have to work its way through the credit assignment problem through brute force.
This is an ongoing development which I hope to make a success story.
Training a physical robot takes a lot of time, and depends on a lot of things. Progress is pretty much halted here. I cannot run training consistently and long enough to get positive results, but the backend is there. The use of N stacked coordinates allowed me to speed up the training program a lot, but I still couldn't get behavior I wanted out of it. This project taught me a lot, and I hope to continue development because I love the Sphero robot, and I think the applications to other robots will be awesome (quadcopter formations, anyone?). If you've read all this, thank you for you time. Here's a brief list of the reasons why I'm pausing here:
- computer instability (training big neural nets can burn up your CPU)
- no option to upload to a more powerful/stable machine
- Bluetooth connection instability
- lack of experience with streaming Bluetooth data
- CV failures, creating reward sparsity and slowing training
- CV errors, creating incorrect rewards and sometimes reversing training
- utility incompatibility (Bluetooth not available for Python on macOS)
I'll continue to develop this as much as possible. I love the Sphero robot and the possibilities it provides to the educational and open source communities. I have begun implementing methods to record and experience replays, and I'm starting to develop a new simulation backend to more rapidly develop basic experiences.
- Python.Swarms: Swarm simulation
- kulka: Sphero Python API
- SKImage: Scikit Learn compatible image processing
- pygame: pygame.camera for webcam image capture
- Keras: Neural network frontend
- theano: Neural network backend
- TensorFlow: Neural network backend
Should run on most Unix (Linux, Mac) but probably not directly on Windows. Coded with Python 3.6 on Mac OS X and Lubuntu 16ish.
- Excellent materials in Georgia Tech's Reinforcement Learning course on Udacity.
- Keras Plays Catch
- Pong from Pixels
- Nervanasys blog post (linked in Karpathy):