Output documentation and examples
QuagmiR is a novel algorithm for isomiR analysis. Each miRNA sequence is divided into three regions: 5’ part, 3’ part and a central region. The latter is unique to each miRNA and will be termed as the “motif” (see more). Reads matching a certain motif are as considered potential isomiRs for the corresponding miRNA (Step 1. Motif pull).
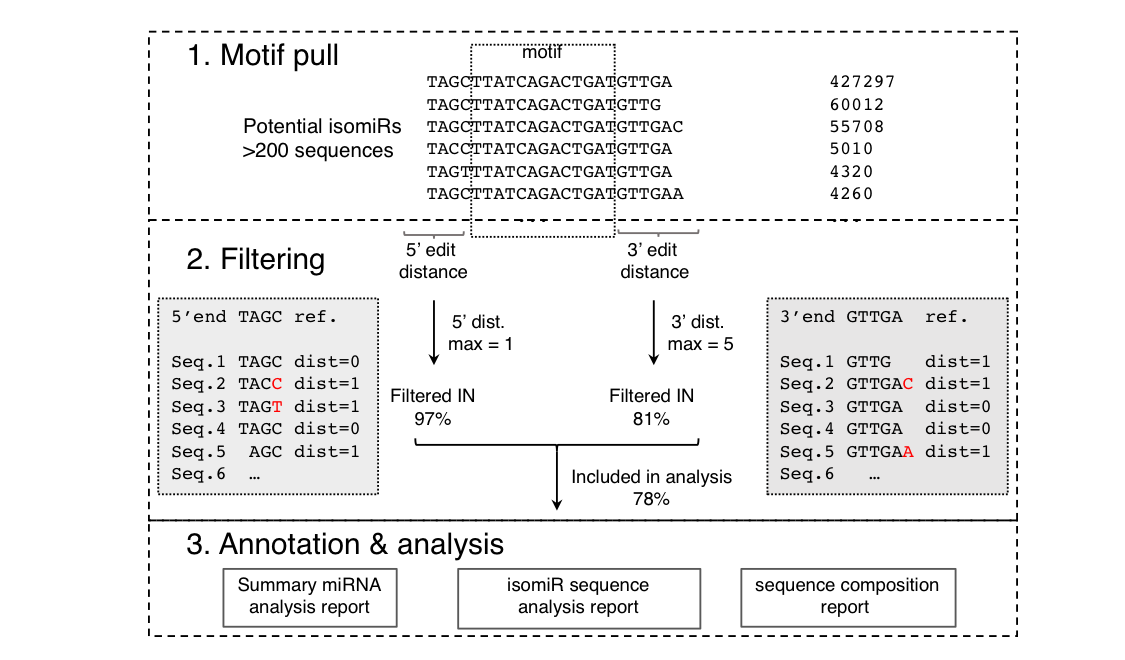
The potential isomiR reads are further filtered according to the nucleotides that precede and follow the motif (5’ part and 3’ part, respectively) (Step 2. Filtering). The sequence pairwise similarity between a read and the reference miRNA is calculated using the Levenshtein distances - the number of deletions, insertions, or substitutions required to transform one string into the other.
Example of Levenshtein distances
Reference sequence: TAGC
Seq.1 TAGC dist=0
Seq.2 TACC dist=1
Seq.3 TAGT dist=1
Seq.4 TAGC dist=0
Seq.5 AGC dist=1
The penalty or distance of any change may be fine-tuned, although the default value is 1. The filtering parameters for the 5' and 3' regions may be set independently to capture the asymmetrical distribution of the sequence heterogeneity. By this approach, users can customize the mapping process to focus on particular types of isomiRs. For example, 3’ isomiRs can be specifically targeted by setting the 5’ distance to 0 and leaving the 3’ distance open to any value (set to -1).
file.collapsed are generated on the first step of analysis and stored under the collapsed folder. They contain the list of all sequences reported on the fastq file and the number of counts for each of them.
7608 TCTTTGGTTATCTAGCTGTATG
14861 TCTTTGGTTATCTAGCTGTATGA
261 TCTTTGGTTATCTAGCTGTATGAA
74 TCTTTGGTTATCTAGCTGTATGAAA
2 TCTTTGGTTATCTAGCTGTATGAAAA
1 TCTTTGGTTATCTAGCTGTATGAAACCA
1 TCTTTGGTTATCTAGCTGTATGAAACCG
1 TCTTTGGTTATCTAGCTGTATGAAAGA
1 TCTTTGGTTATCTAGCTGTATGAAAGGGTTTGA
1 TCTTTGGTTATCTAGCTGTATGAAATA
1 TCTTTGGTTATCTAGCTGTATGAAATCTTGATTTTT
In this example we can see some of the reads pulled with a motif against miR-9.
The report file.isomir.tsv provides the user with a comprehensive list of the isomiRs detected for each reference miRNA. The descriptor provided are the following:
-
MIRNAname of the miRNA -
MOTIFnucleotide sequence used as motif to pull reads -
CONSENSUSmiRNA nucleotide reference sequence (based on miRBase) -
TOTAL_READStotal number of reads for that miRNA after the filtering step -
TOTAL_ISOMIRStotal number of isomiRs detected for that miRNA after the filtering step -
FIDELITY_5P5' isomiRs cleavage fidelity as defined in Gu et al. Cell (2012) -
A_TAILINGpercentage of reads with A in the tail sequence -
C_TAILINGpercentage of reads with C in the tail sequence -
G_TAILINGpercentage of reads with G in the tail sequence -
T_TAILINGpercentage of reads with T in the tail sequence -
SEQUENCE_TRIMMING_ONLYpercentage of reads with trimming, no tail added -
SEQUENCE_TRIMMINGpercentage of reads with trimming -
SEQUENCE_TAILING_ONLYpercentage of reads with tail added on the top of reference sequence -
SEQUENCE_TAILINGpercentage of reads with added tail -
SEQUENCE_TRIMMING_AND_TAILINGpercentage of reads with both trim and tail -
TOTAL_READS_IN_SAMPLEtotal number of reads in the sample (fastq file)
MIRNA MOTIF CONSENSUS TOTAL_READS TOTAL_ISOMIRS FIDELITY_5P A_TAILING C_TAILING G_TAILING T_TAILING SEQUENCE_TRIMMING_ONLY SEQUENCE_TRIMMING SEQUENCE_TAILING_ONLY SEQUENCE_TAILING SEQUENCE_TRIMMING_AND_TAILING TOTAL_READS_IN_SAMPLE
mir-9-5p GGTTATCTAG TCTTTGGTTATCTAGCTGTATGA 16401 502 1.8017 8.7 5.28 59.6 26.42 8.13 18.05 57.75 67.67 9.92 744379
mir-9-3p GCTAGATAAC ATAAAGCTAGATAACCGAAAGT 1321 85 0.1234 16.26 28.53 35.58 19.63 24.21 41.19 1.63 18.61 16.98 744379
The report file.isomir.sequence_info.tsvprovides the user a tabulated description of all the isomiRs detected in a sample and their isomiR features.
-
MIRNAname of the sequence motif that pulled that read -
SEQUENCEsequence of that pulled read -
LEN_READlength (nucleotides) of that pulled read -
READSnumber of counts of that pulled read in the sample -
RATIOpercentage of counts of that particular isomiR sequence relative to the total number of counts considered for that miRNA in the sample -
LEN_TRIMlength (nucleotides) trimmed of that pulled read compared to the reference miRNA -
LEN_TAILlength (nucleotides) tailed of that pulled read compared to the reference miRNA -
SEQ_TAILNucleotide sequence of that pulled read that have been tailed compared to the reference miRNA -
VAR_5Plength (nucleotides) miscleaved by Drosha or Dicer of that pulled read compared to the reference miRNA -
MATCHAny other motif where the pulled read is also a match -
CPMor Counts per Million of that isomiR relative to the total number of reads in the sample -
RPKMof that isomiR relative to the total number of reads in the sample -
DISTANCELevenshtein distances of that particular sequence to the reference miRNA for which it has been selected.
MIRNA SEQUENCE LEN_READ READS RATIO LEN_TRIM LEN_TAIL SEQ_TAIL VAR_5P MATCH CPM RPKM DISTANCE 16401
mir-9-5p TTTGGTTATCTAGCTGTATGAG 22 4774 29.11% 0 1 G 2 269382.6882 291518.1648 3 0 2.664471679
mir-9-5p TTTGGTTATCTAGCTGTATGAGT 23 2063 12.58% 0 2 GT 2 116408.9832 120497.2899 4 1 17.34650326
mir-9-5p TTTGGTTATCTAGCTGTATGA 21 2052 12.51% 0 0 - 2 115788.2857 131269.5357 2 2 78.06841046
mir-9-5p CTTTGGTTATCTAGCTGTATGA 22 1881 11.47% 0 0 - 1 106139.2619 114860.8437 1 3 1.335284434
mir-9-5p TTTGGTTATCTAGCTGTATG 20 594 3.62% 1 0 - 2 33517.66166 39899.02993 3 4 0.268276325
mir-9-5p TTTGGTTATCTAGCTGTATGAA 22 484 2.95% 0 1 A 2 27310.68728 29554.83698 3 5 0.310956649
mir-9-5p TTTGGTTATCTAGCTGTATGAGC 23 282 1.72% 0 2 GC 2 15912.42523 16471.27279 4
mir-9-5p TTTGGTTATCTAGCTGTATGAGA 23 282 1.72% 0 2 GA 2 15912.42523 16471.27279 4
mir-9-5p TTTGGTTATCTAGCTGTATGG 21 231 1.41% 1 1 G 2 13034.6462 14777.41849 3
mir-9-5p TTTGGTTATCTAGCTGTATGAGG 23 217 1.32% 0 2 GG 2 12244.66764 12674.70282 4
mir-9-5p CTTTGGTTATCTAGCTGTATG 21 217 1.32% 1 0 - 1 12244.66764 13881.81737 2
mir-9-5p CTTTGGTTATCTAGCTGTATGG 22 176 1.07% 1 1 G 1 9931.159011 10747.21345 2
mir-9-5p TTTGGTTATCTAGCTGTA 18 148 0.90% 3 0 - 2 8351.201896 11045.74716 5
mir-9-5p TCTTTGGTTATCTAGCTGTATG 22 146 0.89% 1 0 - 0 8238.347816 8915.302066 1
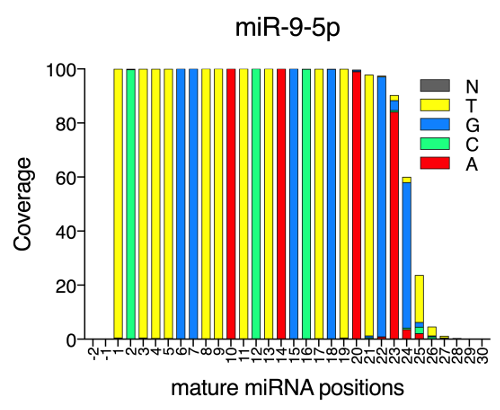
The report file.isomir.nucleotide_dist.tsv provides for each position on the mature miRNA sequence the number of reads covering it and the composition in nucleotides. This allows the user to easily spot any particular position where and editing event takes place or preferred nucleotide tailings by position.
MIRNA NT_POSITION A C G T N READS
mir-9-5p -4 0.0 0.0 0.0 1.0 0.0 1
mir-9-5p -3 0.0 0.0 1.0 0.0 0.0 1
mir-9-5p -2 0.0 0.0 1.0 0.0 0.0 2
mir-9-5p -1 0.75 0.0 0.0 0.25 0.0 4
mir-9-5p 0 0.0022 0.0022 0.0 0.9955 0.0 16395
mir-9-5p 1 0.0003 0.9976 0.0006 0.0015 0.0 16395
mir-9-5p 2 0.0006 0.0036 0.0004 0.9954 0.0 16395
mir-9-5p 3 0.0002 0.0021 0.0003 0.9974 0.0 16395
mir-9-5p 4 0.0004 0.0021 0.0002 0.9973 0.0 16395
mir-9-5p 5 0.0 0.0 1.0 0.0 0.0 16401
mir-9-5p 6 0.0 0.0 1.0 0.0 0.0 16401
mir-9-5p 7 0.0 0.0 0.0 1.0 0.0 16401
mir-9-5p 8 0.0 0.0 0.0 1.0 0.0 16401
mir-9-5p 9 1.0 0.0 0.0 0.0 0.0 16401
mir-9-5p 10 0.0 0.0 0.0 1.0 0.0 16401
mir-9-5p 11 0.0 1.0 0.0 0.0 0.0 16401
mir-9-5p 12 0.0 0.0 0.0 1.0 0.0 16401
mir-9-5p 13 1.0 0.0 0.0 0.0 0.0 16401
mir-9-5p 14 0.0 0.0 1.0 0.0 0.0 16401
mir-9-5p 15 0.0002 0.9988 0.0 0.0009 0.0 16401
mir-9-5p 16 0.0005 0.0017 0.0002 0.9975 0.0 16393
mir-9-5p 17 0.0004 0.0002 0.9988 0.0006 0.0 16390
mir-9-5p 18 0.0006 0.003 0.0007 0.9957 0.0 16389
mir-9-5p 19 0.9935 0.0008 0.0043 0.0015 0.0 16342
mir-9-5p 20 0.0004 0.0055 0.0063 0.9878 0.0 16040
mir-9-5p 21 0.0079 0.0008 0.9878 0.0036 0.0 15975
mir-9-5p 22 0.9316 0.0064 0.0407 0.0212 0.0 14790
mir-9-5p 23 0.0585 0.0097 0.8991 0.0327 0.0 9834
mir-9-5p 24 0.0848 0.1026 0.0735 0.7392 0.0 3880
mir-9-5p 25 0.0519 0.1398 0.0652 0.743 0.0 751
mir-9-5p 26 0.1684 0.1158 0.1368 0.5789 0.0 190
mir-9-5p 27 0.0577 0.1731 0.1731 0.5962 0.0 52
mir-9-5p 28 0.16 0.04 0.28 0.52 0.0 25
mir-9-5p 29 0.1667 0.0 0.1667 0.6667 0.0 12
Example on how the nucleotide composition at base-pair level can be represented.
The group_results allows the user to aggregate results from multiple samples into a single report file. By default the name of this aggregated file is set to cohort1.
An example of this aggregate format looks as follows:
SAMPLE MIRNA MOTIF CONSENSUS TOTAL_READS TOTAL_ISOMIRS FIDELITY_5P A_TAILING C_TAILING G_TAILING T_TAILING SEQUENCE_TRIMMING_ONLY SEQUENCE_TRIMMING SEQUENCE_TAILING_ONLY SEQUENCE_TAILING SEQUENCE_TRIMMING_AND_TAILING TOTAL_READS_IN_SAMPLE
sample1.fastq mir-9-5p GGTTATCTAG TCTTTGGTTATCTAGCTGTATGA 16401 502 1.8017 8.7 5.28 59.6 26.42 8.13 18.05 57.75 67.67 9.92 744379
sample1.fastq mir-9-3p GCTAGATAAC ATAAAGCTAGATAACCGAAAGT 1321 85 0.1234 16.26 28.53 35.58 19.63 24.21 41.19 1.63 18.61 16.98 744379