HowTo
Biopieces comes with a testing suite for testing all (well, only a few for now) Biopieces with all the different options (in a perfect world). Run the following command to test your installation:
bp_test
Biopieces can read configuration information from the ~/.biopiecesrc file. The information in the
file is written as a single Biopiece record, that with respect to default MySQL settings look like
this:
MYSQL_USER: Martin
MYSQL_PASSWORD: Swordfish
---
So, in order to allow the different Biopieces to access the MySQL database to read default username
and password settings - so you don't have to type these out everytime you run one of these
Biopieces - edit the ~/.biopiecesrc on your system with your favorite editor, and enter an entry
like the above and modify the MYSQL_USER and MYSQL_PASSWORD. Finally save the file and change
the permissioins on the file so other users cannot see your password:
chmod 600 ~/.biopiecesrc
If you have a Biopieces pipe that fails there can be a number of different reasons:
- Most often it is misuse where the upstream Biopiece is e.g. outputting the keys
S_IDandQ_IDand the downstream Biopiece is expecting the keySEQ_NAME. - Another catagory of common mistakes is bad data where an empty sequence or a sequence with UIPAC ambiguity codes are used with an external tool that disallow these like
uclust. - Also, it is possible that the data input is truncated or was formatted with carriage returns (i.e. files from Windows).
- There is also computer errors where memory or disk space ran out.
- Finally, you may have encountered a bug in Biopieces - see further down in this section.
In order to locate the error do the following:
- Read the error message carefully - perhaps you forgot a mandatory option or a file was not found.
- Read the wiki pages for the used Biopieces carefully.
- Retry with a few records to minimize the time it takes to reproduce the error. Use the
--numswitch with[read_<format>]. - Retry with the
--verboseswitch to get more information - mostly for wrappers like blast_seq, etc. - Verify the input file format e.g. FASTA and FASTQ can be mistaken.
- Verify the input file's line delimiter i.e. Windows generated text files may have bad line seperators.
- Isolate your problem by dividing the pipe in two and run each seperately. See Howto write the data stream to file
- Repeat 7 for each half.
- Inspect the data stream after each Biopiece in the pipe to verify it contains what you expect.
If the error is not reproducable with a small data set (i.e. using a few records), then there is most likely a problem with the data, like a truncated input file or a missing sequence - or with computer resources like memory or disk space.
If you believe, you have found a bug in Biopieces please report here and please give a reproducable example with a minimal data set.
Often you need to select records from the stream that matches a list of e.g. sequence names. In
order to do this we need grab and a file with a list of identifiers - one per line - that matches
exactly a key in the stream. So to extract particular sequences from e.g. the below FASTA file in
the file test.fna ...
>HA8CWBL01B9XZN
ACGAGTGCGTCCTACGGGAGGCAGCAGTGGGGAATCTTCC
>HA8CWBL01AI903
ATAGAGTACTCCTACGGGGCGCAGCAGTGGGGAATCTTCC
>HA8CWBL01CUX4D
ACGAGTGCGTCCTACGGGGTGCAGCAGTGGGGAATCTTCC
>HA8CWBL01CUSUJ
TGTGAGTAGTCCTATGGGATGCAGCAGTGGGGAATCTTCC
>HA8CWBL01AT53B
CGTCTAGTACCCTACGGGAGGCAGCAGTGGGGAATCTTCC
>HA8CWBL01CERKQ
ACGAGTGCGTCCTACGGGGGGCAGCAGTGGGGAATCTTCC
>HA8CWBL01C382Z
TGTGAGTGGTCCTACGGGACGCAGCAGTGGGGAATCTTCC
>HA8CWBL01AR4X0
TAGTGTAGATCCTACGGGATGCAGCAGTGGGGAATCTTCC
>HA8CWBL01A35I4
ACGAGTGCGTCCTATGGGGTGCAGCAGTGGGGAATCTTCC
>HA8CWBL01C29XM
ACGCGAGTATCCTATGGGAGGCAGCAGTGGGGAATCTTCC
... wanting the records where the SEQ_NAME matches the identifiers in the file test.id:
HA8CWBL01CUX4D
HA8CWBL01AR4X0
HA8CWBL01CUSUJ
HA8CWBL01C29XM
HA8CWBL01A35I4
We simply do:
read_fasta -i test.fna | grab -E test.id -k SEQ_NAME ...
This is easily done using read_fastq. Basically, any entries that contain a
SEQ_NAME and SEQ keys can be written to FASTA files using write_fasta
yields these keys, the conversion is pretty trivial:
read_fastq -i data.fq | write_fasta -o data.fq -x
In order to do this you need to add quality SCORES. This might not be a good idea since you are probably going to feed this bogus quality score data to downstream software that is tuned for real-life data. However, you can simply add quality SCORES using compute to yield a specified score :
read_fasta -i data.fna |
compute -e 'SCORES="I" x SEQ_LEN' |
write_fastq -o data.fq -x
Mapping short sequences to a reference is a two-step process which involves first the indexing of the reference, and second the mapping itself. Biopieces contains wrappers for a number of different short sequence mappers:
And their respective indexing tools:
- create_bowtie_index
- create_bwa_index
- create_usearch_index (not ready yet)
- create_vmatch_index
All of the above pretty much have the same options so it is simply:
read_fasta -i reference.fna |
create_<bowtie|bwa|vmatch>_index -d <directory> -i <index name> -x
The index files will be prefixed with <index name> and then put into the specified <directory>,
which is craeated for the occation.
To map sequences against the index we simply do:
read_fasta -i data.fna |
<bowtie|bwa|vmatch>_seq -i <index name> |
write_bed -o result.bed -x
And the result will be written in BED format. Other formats can be used as well.
BLAT and BLAST are very useful tools for searching long sequences. Biopiece wrappers are available for both:
blat_seq don't require an index, so we can simply do:
read_fasta -i data.fna |
blat_seq -d reference.fna |
write_psl -o result.psl -x
And the result will be written in PSL format. Other formats can be used as well.
BLAST requires an indexed reference sequence and are index in a similar was to the indexes described in [Howto map short sequences].
So for BLAST we do:
read_fasta -i reference.fna |
create_blast_index -d <directory> -i <index name> -x
read_fasta -i data.fna |
blast_seq -d <index name> |
write_bed -o result.bed -x
It is possible to pick the top-5 BLAST hits for each query in a multi-sequence search using index_vals. Below we read in BLAST output saved in tabular format and use index_vals to add an index/forth-running number to each Q_ID in the file. Since BLAST per default outputs the results sorted so the best hits are output first we can then subsequently use grab to pick the first/best 5 hits:
read_blast_tab -i in.blast |
index_vals -k Q_ID |
grab -e 'Q_ID_INDEX <= 5' |
write_blast -o out.blast -x
Searching for patterns is best done with patscan_seq which is a wrapper around scan_for_matches, the best pattern scanning software ever. Patterns can be designed carefully allowing ambiguity codes, mismatches, insertions, and deletions as well as stem loops (nucleotides only). So basically we can do:
read_fasta -i data.fna |
patscan_seq -p <pattern> |
write_bed -o result.bed -x
And the result is written to a BED file.
Now, the pattern can be simple such as a seqeunce: ATCG
Or a more complex pattern like a stem capped by a tetra-loop p1=ATCG 4...4 ~p1[1,0,0] where the one
strand of the stem is ATCG and is assigned to pattern 1 (p1), then followed by a range of four
unspecified residues and finally the reverse complement of p1 allowing for 1 mismatch, 0 insertions and
0 deletions.
So you have a brand new sequence data set and want to get an overview ...
The first thing is to figure out what type of file format the data is store in.
FASTA data are typically stored in files with .fna, .fa, .faa, or .fasta suffixes and these
can be read using read_fasta. FASTQ files typically have .fq or .fastq suffixes and can be read
using read_fastq. Data from Roches's 454 platform can come as SFF with .sff suffix and can be read
with read_sff, but can also come in two files, one a FASTA file with the sequence and a FASTA like
file with the quality scores. These can be read with [read_454]. And now to the basic questions:
To find out how many sequences you have use count_records:
read_fasta -i data.fna | count_records -x
Which will output a record with the sequence count:
RECORDS_COUNT: 12345678
---
A bit more advanced which in addtion to record count will tell you the minimum, maximum and mean sequence lengths among other things:
read_fasta -i data.fna | analyze_vals -x | write_tab -cCpx
Then you get a table like this:
+----------+------------+--------+-----+-------+-----------+-------+
| KEY | TYPE | COUNT | MIN | MAX | SUM | MEAN |
+----------+------------+--------+-----+-------+-----------+-------+
| SEQ_NAME | Alphabetic | 10,000 | 37 | 41 | 398,895 | 39.89 |
| SEQ | Alphabetic | 10,000 | 120 | 1,108 | 4,810,958 | 481.1 |
| SEQ_LEN | Numeric | 10,000 | 120 | 1,108 | 4,810,958 | 481.1 |
+----------+------------+--------+-----+-------+-----------+-------+
Which is basically the output of these Biopieces:
If you want to analyze the sequence composition you can use analyze_seq which will yield the residue composition, and the percentages of how many residues are soft masked (i.e. lower case) and hard masked (i.e. N's), and the GC%.
SEQ_NAME: ILLUMINA-52179E_0004:2:1:1040:5263#TTAGGC/1
SEQ: TTCGGCATCGGCGGCGACGTTGGCGGCGGGGCCGGGCGGGTCGANNNCAT
SEQ_LEN: 50
RES[[T]]: 7
RES[[C]]: 13
RES[[G]]: 23
RES[[A]]: 4
RES[[N]]: 3
SOFT_MASK%: 0.0
HARD_MASK%: 6.0
GC%: 72.0
---
Thus to get the mean HARD_MASK% for all records in the stream, you need to use analyze_seq followed by mean_vals like this:
read_fasta -i data.fna |
analyze_seq |
mean_vals -k HARD_MASK% -x
Which outputs the mean in a record like this:
HARD_MASK%_MEAN: 3.80
REC_TYPE: MEAN
---
See also Howto determine the GC content of a data set.
Plotting the length distribution of a data set involves reading in the data and piping it into plot_distribution:
read_fasta -i data.fna |
plot_distribution -k SEQ_LEN -t png -o plot.png -x
And the plots look like this:
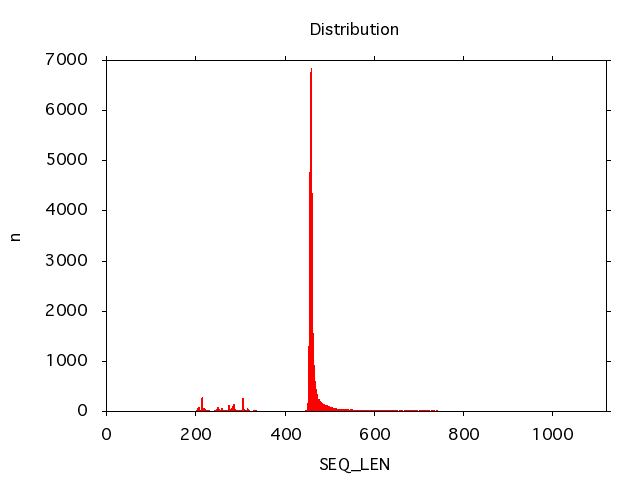
It is possible to detect problems with the nucleotide distribution of shotgun sequence data using plot_nucleotide_distribution:
read_fastq -i data.fq |
plot_nucleotide_distribution -t png -o plot.png -x
And the plots look like this:
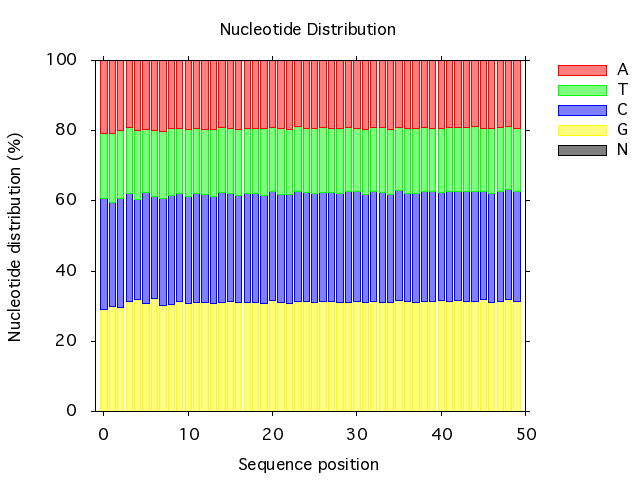
To get the mean GC content of a sequence you can use analyze_gc, and if you want the mean GC content for a stack of sequences you simply calculate the mean using mean_vals:
read_fasta -i data.fna |
analyze_gc |
mean_vals -k GC% -x
Which will output a record with the mean GC content:
GC%_MEAN: 37.50
---
A distribution of the GC content can be created like this:
read_fasta -i test.fna |
analyze_gc |
bin_vals -b 5 -k GC% |
plot_distribution -k GC%_BIN -o gc_dist_plot.png -t png -x
To get the following plot:
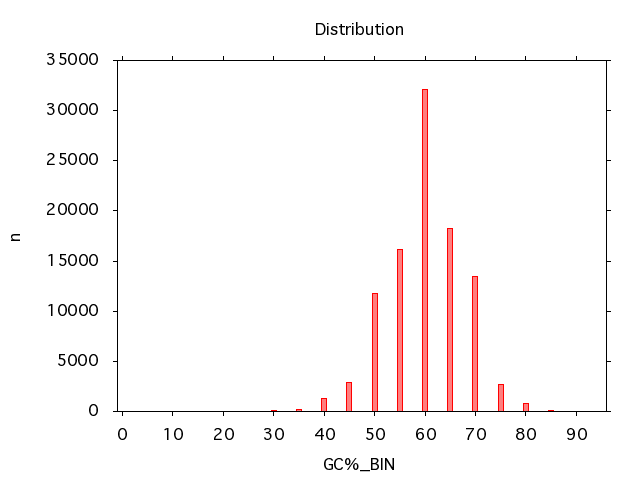
With the comming of the Illumina HiSeq platform the sequence data from each experiment may come as multiple files. These can be collected like this:
read_fastq -i ‘experiment_X_*.fastq.gz’ |
write_fastq -Zo experiment_X_all.fq.gz -x
Notice that we keep the data zipped to avoid filling up the disks unnecessarily.
To filter sequences where the length is outside a given interval e.g. 100-200, we can use grab twice:
read_fastq -i in.fq |
grab -e 'SEQ_LEN >= 100' |
grab -e 'SEQ_LEN <= 200 |
write_fastq -o out.fq -x
Some sequences have suboptimal quality scores and have been flagged as filtered by the CASAVA 1.8 pipeline. These contain a Y in the sequence name as in "filtered? Yes!". We need to remove these like this:
read_fastq -i data.fq.gz |
grab -ip ':Y:' -k SEQ_NAME |
write_fastq -Zo data_filt.fq.gz -x
It is probably a good idea to check how many sequences were filtered. Use count_records for that.
Barcodes are DNA tags of 6-11 bases that are added to experiements allowing for simultaniously sequencing of multiple experiments, which can then be seperated after sequencing based on the barcodes. Splitting a NGS data set on DNA barcodes can be done by reading the data followed by find_barcodes to locate (and remove) the barcodes, and finally, write the sequences to files according to the barcode name with write_fasta_files like this:
read_sff -i data.sff | # Read the data from a SFF file
find_barcodes -p 4 -m 2 | # Look for barcode at position 4 and allow for 2 mismatches
write_fasta_files -d Output_dir -k BARCODE_NAME -x # Write FASTA files
And the resulting files will end up in the Output_dir:
Output_dir/
|-- MID1.fasta
|-- MID10.fasta
|-- MID104.fasta
|-- MID106.fasta
|-- MID107.fasta
|-- MID109.fasta
...
In the above example we are looking for Roche's MID barcodes, which are hard coded in find_barcodes. It is possible to use a list of custom barcodes ).
It is possible to start with FASTA and QUAL files using [read_454], and it is possible to preserve the quality scores by saving the output in FASTQ format using write_fastq_files.
Plot the mean quality score using plot_scores like this:
read_fastq -i data.fq.gz |
plot_scores -t png -o data_scores.png -x
This will output a PNG file with the mean scores:
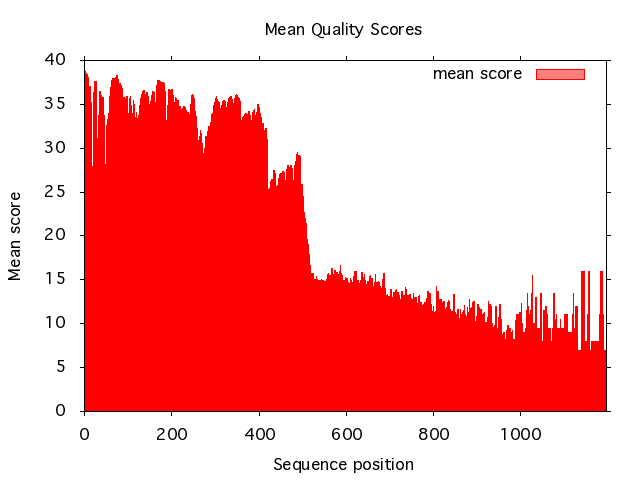
Since the quality scores of the sequences deteriorate with sequence length, we need to trim sequences from the ends that have suboptimal quality scores. We do this pretty conservatively by using the threshold Q20 with trim_seq. Also, since some sequences will be trimmed down to length 0, we need to filter these ) an only keep sequences of a minimum length:
read_fastq -i data.fq.gz |
trim_seq -m 20 |
grab -e ‘SEQ_LEN>=50’|
write_fastq -Zo data_etrim.fq.gz -x
Some sequences may contain a region with suboptimal sequence after end trimming (see Howto trim sequence ends). These can be identified by running a sliding window over the sequence to locate local minima, and if any such is found, the sequence can be discarded. We use mean_scores:
read_fastq -i Ex1_filt_etrim.fq.gz |
mean_scores -l |
grab -e ‘SCORES_MEAN_LOCAL >= 15’ |
write_fastq -Zo Ex1_filt_etrim_ltrim.fq.gz -x
Do count how many sequences were filtered using count_records. Also you can plot the mean scores. See Howto plot quality scores.
Adaptors can be located with find_adaptor that scan sequences for given adaptor sequences allowing for a specified percentage of mismatches, insertions and deletions. It can also locate partial adaptors that are often found at the sequence ends. Next the located adaptors can be removed with clip_adaptor.
read_fastq -i test.fq |
find_adaptor -l 1 -L 1 -f ACACGACGCTCTTCCGATCT -r TCTAGCCTTCTGTGTGCAGA |
clip_adaptor |
write_fastq -o test_noAdaptor.fq -x
Pair-end sequences are generally output in correct order from the sequencing instruments, and to maintain that order with Biopieces it is possible to read in the data from two list of files using read_fastq:
read_fastq -i pair1.fq -j pair2.fq |
write_fastq -o interleaved.fq
Interleaved records in the stream are required for assembly with assemble_pairs, and assemble_seq_velvet, etc.
If, for some reason, the order of pairs is lost it is possible to restore the order with order_pairs, however, it is much more efficient - time and memory wise - to make sure the original order is kept.
Cleaning pair-end sequences is a two step process:
- Interleaving sequence.
- Actual trimming and filtering.
The trimming part involves end-trimming and adaptor removal, while the filtering part utilizes a trick where the paired sequences are merged into a single sequence while the length of the left and right sequence is recorded. Thus, it is possible to filter on the left or right seqeunce length and, in the merged sequence, filter sequences with local areas of suboptimal quality. Finally, the merged sequences are split and:
read_fastq -i interleaved.fq |
trim_seq -m 25 -l 3 |
find_adaptor -l 6 -L 6 -f ACACGACGCTCTTCCGATCT -r AGATCGGAAGAGCACACGTC |
clip_adaptor |
merge_pair_seq |
grab -e 'SEQ_LEN_LEFT >= 30' |
grab -e 'SEQ_LEN_RIGHT >= 30' |
mean_scores -l |
grab -e 'MEAN_SCORE_LOCAL >= 15' |
split_pair_seq |
write_fastq -o cleaned.fq -x
See also Example processing pair-end FASTQ files.
Assembly is the art of stitching together short sequences into longer contig -uous regions. Genome assembly without a reference to use as backbone is called de novo assembly. De novo assembly using 454 reads should be done with Newbler and MIRA for the best results.
Illumina reads can be assembled using the following Biopieces which are basic wrappers:
There is no single best way of performing genome assembly, so it is recommended to do a number of assemblies with different filtering and assemblers and compare the results. The assembler Biopieces do multiple assemblies with a range of different parameters, like k-mer sizes. Also, the starting data may or may not benefit from rigorously trimming and cleaning.
So for all of the assemblers you can do this:
read_fastq -i data.fq.gz |
assemble_seq_<assembler> -d <directory> -k <min k-mer size> -K <max k-mer size> |
write_fasta -o contigs.fna -x
The assemble_seq_velvet will produce a number of assemblies that will be
stored in the <directory> and only the best assembly - determined from the highest N50 - will be
output to the stream and stored in the FASTA file. assemble_seq_idba is iterative and only outputs
the best assembly.
The result of the assemblies can be evaluated using analyze_assembly (see Howto analyze assembled contigs).
Assembly is done using Meta-IDBA.
read_fastq -i Ex1_filt_etrim_ltrim.fq.gz |
assemble_seq_idba -m -k 50 -K 80 -d IDBA |
write_fasta -Zo Ex1_assembly.fna.gz
metavelvet - needs to be studies (probably in Japanese).
Assembled contigs can be analyzed to get some stats using analyze_assembly. We include a filtering step to discard contigs shorter than 200 bases:
read_fasta -i contigs.fna |
grab -e "SEQ_LEN>=200" |
analyze_assembly -x
And the output:
N50: 9082
MAX: 52038
MIN: 200
MEAN: 4170
TOTAL: 3057214
COUNT: 733
---
Notice that with the -g switch analyze_seq will calculate a predicted gene coverage that is a
useful metric for comparing multiple assemblies.
Many different tools exists for calling single nucleotide polymorphims (SNP) and deletion/insertion polymophisms (DIP). Basically, these work by mapping sequences to a reference and do advanced statistics on the descrepancies. Sometimes, this is follwed by realignment sequences around the positions of interest - and more statistics. Biopieces have a naive step-wise method which focusses on rigorous trimming and cleaning of sequnces to minimize the impact of SNPs and DIPs caused by residues with substandard quality. So the steps are:
- Trim sequences.
- Filter sequences with substandard qualities.
- Map sequences to reference.
- find_SNPs
SNPs and DIPs can be found in KISS or SAM type records using find_SNPs:
read_sam -i mapping_result.sam |
find_SNPs |
grab -e "REC_TYPE eq SNP" |
sort_records -rk SNP_COUNTn |
write_tab -cx
Which will yield a table outlined below, from where it is easy to distinguish real SNPs from the
SNP_COUNT alone:
#SNP_COUNT EVENT S_ID REC_TYPE TYPE POS
65 T>G gi|48994873|gb|U00096.2| SNP MISMATCH 2500
62 G>- gi|48994873|gb|U00096.2| SNP DELETION 7000
57 T>A gi|48994873|gb|U00096.2| SNP MISMATCH 2000
55 C>A gi|48994873|gb|U00096.2| SNP MISMATCH 1500
51 G>C gi|48994873|gb|U00096.2| SNP MISMATCH 1000
49 G>- gi|48994873|gb|U00096.2| SNP DELETION 7500
48 T>G gi|48994873|gb|U00096.2| SNP MISMATCH 500
43 ->A gi|48994873|gb|U00096.2| SNP INSERTION 3500
36 ->A gi|48994873|gb|U00096.2| SNP INSERTION 2999
4 T>A gi|48994873|gb|U00096.2| SNP MISMATCH 3499
All Biopieces write the data stream to 'stdout' by default which can be written to a file by
using the ubiquitous -O switch:
<biopiece> -O <file>
It is also possible to use redirection <biopiece> > <file>, but beware that if the biopiece outputs
data e.g. FASTA entries then the FASTA data and Biopiece data will be mixed and causing havock.
If you want to read a data stream that you previously have saved to file in Biopieces format (see Howto write the data stream to file) do:
<biopiece> -I <file>
This switch works with all Biopieces. It is also possible to read in data from multiple sources by repeating:
<biopiece> -I <file1> | <biopiece> -I <file2>
It is also possible to use a glob expression:
<biopiece> -I "*.stream"
Note that the glob expression must be in "".
It is also possible to use redirection with cat <file> | <biopiece> or <biopiece> < <file>.
It is also possible to read in a saved Biopieces stream ) as well as reading data in one go:
<biopiece> --stream_in=<file1> --data_in=<file2>
If you want to read data from several files you can do this:
<biopiece> --data_in=<file1> | <biopiece> --data_in=<file2>
If you have several data files you can read in all explicitly with a comma separated list:
<biopiece> --data_in=file1,file2,file3
And it is also possible to use file globbing:
<biopiece> --data_in="*.fna"
Or in a combination:
<biopiece> --data_in="file1,/dir/*.fna"
Finally, it is possible to read in data in different formats using the appropriate Biopiece for each format:
<biopiece1> --data_in=<file1> | <biopiece2> --data_in=<file2> ...
It is possible to utilize the bash shell to loop over multiple input files. If for an example you have multiple experiments each in one FASTA file, and you for all the files want to obtain the sequences longer than 100 in a new file, you can do:
for i in *.fna;
do read_fasta -i $i | grab -e 'SEQ_LEN>=100' | write_fasta -o `basename $i .fna`._gt100.fna -x;
done
Notice that we use the shell tool basename to rename the output files from my_experiment.fna to
my_experiment_gt100.fna.
You can make aliases to popular commands to save time typing:
alias revcomp_seq="reverse_seq | complement_seq"
Which makes it possible to reverse-complement sequences like this:
read_fasta -i data.fna | revcomp_seq | write_fasta -o data_rc.fna -x
You can also use aliases as shortcuts to Biopieces with specific arguments which saves typing:
alias blast_refseq="blast_seq -d /var/databases/blast/refseq"
To save aliases from session to session save these in your .bashrc file.
Often you get a pipeline that is quite long and to avoid re-typing the commands and avoid the hazzle of copy/pasting across several lines the easy way is to put the pipeline in a shell script. To do this, open a text file with your favorite text editor and enter something like this:
#!/bin/sh
read_fasta -i HPV.fna | # read in the Human Papilloma Virus genome
add_ident -k SEQ_NAME | # add a new identifier to all sequences
indel_seq -i 1 -d 1 | # add 1 insertions and 1 indel to each sequence
mutate_seq -n 1 | # mutate 1 residue in the sequence
write_fasta -o HPV_mutant -x # write sequences to file.
The first line #!/bin/sh is the hashbang or shebang line that tells the operating system that
this file should be interpreted with the sh or shell command. Note that you can have comments in
the Biopiece script using #. After saving the file (here we saved it as test.sh) you can change
the permissions on the file to make it executable so it may be run from the commandline:
chmod "+x" test.sh
./test.sh
Another use for scripting Biopieces is to get a clear seperation of Biopieces code and everything else. Such a script would be reading raw data of a particular format from STDIN and write raw data of a particular format to STDOUT. E.g.
#!/bin/sh
read_fastq -i - | # read raw FASTQ data from STDIN
grab -ip ':Y:' -k SEQ_NAME | # Grab all records wihtout :Y: in the sequence name
write_fastq -x # write raw FASTQ data to STDOUT
Now this can be executed from the command line and we observed that the Biopieces part is abstracted away:
cat data_in.fq | ./test.sh > data_out.fq
Biopiece records are based on hash type structures with key/value pairs on separate lines
(terminated with a newline or \n) using : (colon followed by a white space) to assign the
right side value to the left side key: key: value. Records are delimited by lines of ---. This
simple type of records has a number of significant advantages over more complex hierarchical
structures such as YAML or XML:
- speed Biopiece records are much faster to parse and emit than more complex data types.
- simplicity a simple type of data record is more understandable for users, and easier to manipulate for developers, remember that not everyone is an expert programmer.
- compatibility simple records allow for easy compatability between Biopieces, but also with standard UNIX tools, sed, and awk.
However, since there are very little restaints in the Biopiece format, it is possible to use complex records by serializing the data into strings.
One example of complex data that is used in a number of Biopieces are a list of elements, such as the quality SCORES in read_fastq:
SCORES: 40;40;40;40;40;40;40;40;40;40;40;40
This is simply a list of scores seperated by a delimiter that in this case is ; and which easily
can be used to split the different values. There is no restraints as to how a simple list like this
is defined. It could be in brackets and separated by commas to follow the Perl data structure
syntax, or it could be something completely different - it is up to the Biopieces that manipulates
the relevant data.
If you as an example want to use a list of lists you can assign it to a Biopiece records as demonstrated with this table:
A1 B1 C1
A2 B2 C2
A3 B3 C3
Which can be written as a Biopiece record like this:
cat test.stream
TABLE: [ [ 'A1', 'B1', 'C1' ], [ 'A2', 'B2', 'C2' ], [ 'A3', 'B3', 'C3' ] ]
---
Now, this way of serializing the table is based on Perl data structures where [] denotes a list
of comma separated elements, and embedded [[],[],[]] denotes a list of lists. The advantages here
is that we can use Perl's eval command to allow the Perl parser to quickly translate the string
to a Perl data structure. We do this with bioscript:
bioscript -I test.stream -e '$r = get_record( $in ); $r->{ TABLE2 } = eval $r->{ TABLE }; print Dumper( $r )'
$VAR1 = {
'TABLE' => '[ [ \'A1\', \'B1\', \'C1\' ], [ \'A2\', \'B2\', \'C2\' ], [ \'A3\', \'B3\', \'C3\' ] ]',
'TABLE2' => [
[
'A1',
'B1',
'C1'
],
[
'A2',
'B2',
'C2'
],
[
'A3',
'B3',
'C3'
]
]
};
As you can see, TABLE2 contains the eval'ed data which now can be manipulated the Perl way.
Of cause we cauld have done the eval in place and thus avoid creating a TABLE2 key:
bioscript -I test.stream -e '$r = get_record( $in ); $r->{ TABLE } = eval $r->{ TABLE }; print Dumper( $r )'
$VAR1 = {
'TABLE' => [
[
'A1',
'B1',
'C1'
],
[
'A2',
'B2',
'C2'
],
[
'A3',
'B3',
'C3'
]
]
};
Similarly, different XML or YAML parsers can be can be used instead of eval, however, it is up to
you to create the Biopieces that emits, parses, and manipulates complex Biopiece records.
Only use complex records if you really must. Often it is better to rethink the approach so it can be handled with the ordinary Biopiece records format.
For some tasks it is possible to use GNU Parallel which allow execution of Biopieces in parallel using multiple CPUs on multiple computers.
In brief, GNU Parallel works by reading from a pipe the content of e.g. a FASTA file which is then divided into chunks for which
a Biopieces pipeline is executed on a separate CPU with the output written to stdout so that it may be redirected to an output
file.
Here is an example where we use GNU Parallel to process a list of FASTA files:
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test1.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test2.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test3.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test4.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test5.fna
parallel 'read_fasta -i {} | extract_seq -l 5 | write_fasta -o {.}_trim.fna -x' ::: *.fna
And the result:
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test1.fna
-rw-r--r-- 1 maasha users 558 Sep 12 14:00 test1_trim.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test2.fna
-rw-r--r-- 1 maasha users 558 Sep 12 14:00 test2_trim.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test3.fna
-rw-r--r-- 1 maasha users 558 Sep 12 14:00 test3_trim.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test4.fna
-rw-r--r-- 1 maasha users 558 Sep 12 14:00 test4_trim.fna
-rw-r--r-- 1 maasha users 1395 Sep 12 12:25 test5.fna
-rw-r--r-- 1 maasha users 558 Sep 12 14:00 test5_trim.fna
In the below example we use the power of GNU Parallel to grab in a huge FASTQ file, we use cat and read_fastq -i - where the
use of -i - which allows read_fastq to read raw FASTQ data from stdin to parallel which gives a solid performance boost
when using parallels --pipe switch and -L 4 to indicate a FASTQ entry of four lines. This allows parallel to correctly
divide the raw FASTQ stream into even sized chunks. Following all the arguments is the Biopieces pipeline that we want to run -
and the result is collected in out.fq:
NB this requires parallel version >= 20130222 - or it will be very slow
cat in.fq |
parallel --block 100M -k --pipe -L 4 "read_fastq -i - | grab -k SEQ_NAME -p ':Y:' | write_fastq -x"
> out.fq
To setup a complete Biopieces pipeline for heavy duty tasks it is best done by writing a shell script with the Biopieces commands and execute this script using GNU Parallel.
Here is an example script illumina_trim_filt.sh that does quality trimming and filtering of Illumina data:
#!/bin/sh
read_fastq -e base_33 -i - | # Read in raw FASTQ data from stdin.
grab -ip ":Y:" -k SEQ_NAME | # Filter reads flagged by CASAVA.
find_adaptor -l 1 -L 1 -f ACACGACGCTCTTCCGATCT -r AGATCGGAAGAGCACACGTCT | # Find adaptors including partial adaptors.
clip_adaptor | # Clip found adaptors.
trim_seq | # End trim sequences.
grab -e 'SEQ_LEN >= 50' | # Filter reads shorter than 50 bp.
mean_scores | # Calculate mean quality score.
grab -e "SCORES_MEAN >= 20" | # Filter reads with score mean below 20.
mean_scores -l | # Calculate local mean quality score.
grab -e "SCORES_MEAN_LOCAL >= 15" | # Filter reads with local score mean below 15.
write_fastq -x # Write raw FASTQ data to stdout.
Save the script in a file illumina_trim_filt.sh and make it executable:
chmod +x illumina_trim_filt.sh
Notice that this script reads and writes raw FASTQ data and thus the Biopieces part is abstracted away.
Now the script can be tested:
cat data.fq | illumina_trim_filt.sh > data_trim_filt.fq
And parallized:
parallel --pipe --block 100M -k -L 4 'cat {} | illumina_trim_filt.sh > {.}_trim_filt.fq' ::: *.fq
First we need to Interleaving the sequences see Howto interleave pair-end sequences.
Next we replace the illumina_trim_filt.sh script in [Example processing multiple FASTQ files] with the following:
#!/bin/sh
read_fastq -e base_33 -i - | # Read in interleaved FASTQ data from stdin.
trim_seq -m 25 -l 3 | # End trim sequences.
find_adaptor -l 6 -L 6 -f ACACGACGCTCTTCCGATCT -r AGATCGGAAGAGCACACGTC | # Find adaptors including partial adaptors.
clip_adaptor | # Clip found adaptors.
merge_pair_seq | # Merge sequence pairs.
grab -e 'SEQ_LEN_LEFT >= 30' | # Filter reads shorter than 30 bp.
grab -e 'SEQ_LEN_RIGHT >= 30' | # Filter reads shorter than 30 bp.
mean_scores -l | # Calculate local mean quality score.
grab -e 'SCORES_MEAN_LOCAL >= 15' | # Filter reads with local score mean below 15.
split_pair_seq | # Split sequence pairs.
write_fastq -x # Write interleaved FASTQ data to stdout.
Finally, we can feed the interleaved data to parallel making sure we split in blocks with an equal amount of reads using -L 8:
cat interleaved.fq | parallel --pipe --keep-order --block 50M -L 8 ./illumina_trim_filt.sh > clean.fq
See also Howto clean pair-end sequences.
The usage information for the Biopieces is the help text you get when running a Biopiece with no arguments, or if you are browsing the Biopieces online on the wiki http://code.google.com/p/biopieces/w/list. The usage information is written in Google wiki format http://code.google.com/p/support/wiki/WikiSyntax which means that the usage information is rendered nicely in HTML on the wiki (e.g. [http://code.google.com/p/biopieces/wiki/align_seq align_seq]) and a specific Biopieces print_wiki renders a nice ASCII-text version. This means that you as a developer only need to call the print_wiki Biopiece to render the usage information independent of programming language:
print_wiki [options] -i <usage file.wiki>
See print_wiki for more details.
The usage information exists in one wiki file per Biopiece. For an example the wiki file for the align_seq is located here:
~/biopieces/wiki/align_seq.md
or using the $BP_DIR environment variable:
$BP_DIR/wiki/align_seq.md
The basename of the wiki page must be the same as the Biopieces i.e. align_seq for the above
example.
When creating a new Biopieces start by copying an existing wiki file and edit the content. Do not change the structure of the wiki file - all the sections are mandatory.
Do try to re-use the option names - both the full option name and and the single character name.
The argument options are devided into different catagories that are used for argument checking:
- flag - the option is a flag and does not take an argument.
- string - the argument is a string.
- list - the argument is a comma separated string with no whitespace.
- int - the arguemnt is a integer - both signed and unsigned including 0.
- uint - the argument is a unsigned integer i.e. only positive integers including 0.
- float - the arguemnt is a decimal number.
- file - the argument is a file.
- file! - the arguemnt is a file that must exist.
- files - the argument is a comma separated list of files or file glob expressions..
- files! - same as the above, but the resulting files must all exist.
- dir - the argument is a directory.
- dir! = the argument is a directory that must exist.
- genome - the argument is a genome that must be formatted with format_genome).
Remember that there are four ubiquis option names - both long and short - that are reserved:
[-? | --help] # Print full usage description.
[-I <file> | --stream_in=<file!>] # Read input stream from file - Default=STDIN
[-O <file> | --stream_out=<file>] # Write output stream to file - Default=STDOUT
[-v | --verbose]
Note that flag type options are left blank in the wiki files.
First copy and modify an existing wiki page, see Howto write usage information.
Use the code from a related Biopiece as template if possible, or use one of the simple ones such as split_vals as template.
cp $BP_BIN/split_vals $BP_BIN/<new Biopiece>
After stripping down the part of the code omitting the parts that were specific to the function of split_seq we end up with this very basic Biopiece (without the line numbering):
1 #!/usr/bin/env perl
2
3 # Copyright (C) 2007-2009 Martin A. Hansen.
4
5 # This program is free software; you can redistribute it and/or
6 # modify it under the terms of the GNU General Public License
7 # as published by the Free Software Foundation; either version 2
8 # of the License, or (at your option) any later version.
9
10 # This program is distributed in the hope that it will be useful,
11 # but WITHOUT ANY WARRANTY; without even the implied warranty of
12 # MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
13 # GNU General Public License for more details.
14
15 # You should have received a copy of the GNU General Public License
16 # along with this program; if not, write to the Free Software
17 # Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301, USA.
18
19 # http://www.gnu.org/copyleft/gpl.html
20
21
22 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>> DESCRIPTION <<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
23
24 # Split the values of a key into new key/value pairs.
25
26 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
27
28 use warnings;
29 use strict;
30 use Maasha::Biopieces;
31
32
33 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
34
35
36 my ( $options, $in, $out, $record, @vals, $i );
37
38 $options = Maasha::Biopieces::parse_options(
39 [
40 { long => 'key', short => 'k', type => 'string', mandatory => 'yes', default => undef, allowed => undef, disallowed => undef },
41 { long => 'keys', short => 'K', type => 'list', mandatory => 'no', default => undef, allowed => undef, disallowed => undef },
42 { long => 'delimit', short => 'd', type => 'string', mandatory => 'no', default => '_', allowed => undef, disallowed => undef },
43 ]
44 );
45
46 $in = Maasha::Biopieces::read_stream( $options->{ "stream_in" } );
47 $out = Maasha::Biopieces::write_stream( $options->{ "stream_out" } );
48
49 while ( $record = Maasha::Biopieces::get_record( $in ) )
50 {
51 Maasha::Biopieces::put_record( $record, $out );
52 }
53
54 Maasha::Biopieces::close_stream( $in );
55 Maasha::Biopieces::close_stream( $out );
56
57
58 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
59
60
61 BEGIN
62 {
63 Maasha::Biopieces::status_set();
64 }
65
66
67 END
68 {
69 Maasha::Biopieces::status_log();
70 }
71
72
73 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
74
75
76 __END__
Let us go over the different sections in this minimalistic Biopiece:
Line 1. First we have the hashbang line #!/usr/bin/env perl, note that we use env perl and not a fixed path!
Lines 3-19. Next we have the license text.
Lines 22-26. This section contains a short description that typically is a copy of the Summary section from the wiki file.
Lines 28-30. Here we have a section with use statements. We always have the use warnings and
use strict pragmas enabled in Perl, and the use Maasha::Biopieces; enables subroutines from
this module, that also always is included. You can include other modules as well. A lot of useful
existing modules can be located in $BP_PERL or ~/biopieces/code_perl/. The best way to locate
useful modules is to look at the modules included by related Biopieces.
Lines 38-44. In this section the Maasha::Biopieces::parse_options subroutine call is of
particular interest since this contains the information on the options and their arguments. The
argument to the subroutine is a list of hash references. In this particular example the Biopiece
have three options apart from the mandatory 4 options that are included in all Biopieces:
The three options specific to the split_vals Biopiece:
$options = Maasha::Biopieces::parse_options(
[
{ long => 'key', short => 'k', type => 'string', mandatory => 'yes', default => undef, allowed => undef, disallowed => undef },
{ long => 'keys', short => 'K', type => 'list', mandatory => 'no', default => undef, allowed => undef, disallowed => undef },
{ long => 'delimit', short => 'd', type => 'string', mandatory => 'no', default => '_', allowed => undef, disallowed => undef },
]
);
The four mandatory options added to the $options hash reference when calling
Maasha::Biopieces::parse_options:
{ long => 'help', short => '?', type => 'flag', mandatory => 'no', default => undef, allowed => undef, disallowed => undef },
{ long => 'stream_in', short => 'I', type => 'file!', mandatory => 'no', default => undef, allowed => undef, disallowed => undef },
{ long => 'stream_out', short => 'O', type => 'file', mandatory => 'no', default => undef, allowed => undef, disallowed => undef },
{ long => 'verbose', short => 'v', type => 'flag', mandatory => 'no', default => undef, allowed => undef, disallowed => undef },
The different keys are:
- long The long option name - do reuse sensible names from other Biopieces!
- short The short option name - do reuse sensible names from other Biopieces!
- type See the type explanations in the [Howto write usage information] section.
- mandatory Indicate if the option must have an argument.
- default Allow a default value or a comma seperated string of values depending on the type.
- allowed Allow a comma seperated string of allowed arguments.
- disallowed A comma seperated string of arguments not allowed.
If you need more advanced argument checking than the parse_options subroutine provides, you will
have to do it yourself after parse_options is called. For an example:
Maasha::Common::error( qq(both --database and --genome specified) ) if $options->{ "genome" } and $options->{ "database" };
Maasha::Common::error( qq(no --database or --genome specified) ) if not $options->{ "genome" } and not $options->{ "database" };
If your Biopiece does not need any non-mandatory options you just call parse_options with no
arguments like this:
$options = Maasha::Biopieces::parse_options();
Lines 46-47. Here we open filehandles to incomming data from file or STDIN and outgoing data to file or STDOUT.
Lines 49-52. Here we have the main iterative loop of the Biopiece. You can manipulate the $record
hash references from the data stream.
Lines 54-55. Here we close the filehandles to the data stream.
Lines 61-70. The BEGIN section sets the status file before anything else happens. The END section reads the status file and at the end of the Biopiece execution write the Biopiece log entry to the log file.
If you want to modulate your code, which is always a good idea, you can place subroutines in a
section beginning at line 57. Or better still. Write a Module and place in
~/biopieces/code_perl/<your_username>/ and include this module in the include section lines
29-30.
If you need temporary files you can get a temporary directory using the subroutine
Maasha::Biopices::get_tmpdir() that creates a directory in the $BP_TMP directory based on
username and session id. The path to this temporary directory is returned, and the directory is
automagically destroyed when the Biopieces execution is done - also, if the Biopiece is interupted
the now stale temporary directory will be cleaned out when any other Biopiece is executed.
$tmp_dir = Maasha::Biopieces::get_tmpdir();
Use the verbose option to allow for messages to show progress or debug:
$count = 1;
while ( $record = Maasha::Biopieces::get_record( $in ) )
{
Maasha::Biopieces::put_record( $record, $out );
print STDERR "Records processed: $count\n" if $options->{ 'verbose' } and $count % 1000 == 0;
$count++;
}
Finally, add some tests to the Biopieces testing suite to check that everything is working. See Howto write tests for the Biopieces test suite.
First copy and modify an existing wiki page, see Howto write usage information.
Use the code from a related Biopiece as template if possible, or use one of the simple ones such as swapcase_seq as template.
cp $BP_BIN/swapcase_seq $BP_BIN/<new Biopiece>
The swapcase_seq Biopiece is very simple and the code is showed below:
1 #!/usr/bin/env ruby
2
3 # Copyright (C) 2007-2011 Martin A. Hansen.
4
5 # This program is free software; you can redistribute it and/or
6 # modify it under the terms of the GNU General Public License
7 # as published by the Free Software Foundation; either version 2
8 # of the License, or (at your option) any later version.
9
10 # This program is distributed in the hope that it will be useful,
11 # but WITHOUT ANY WARRANTY; without even the implied warranty of
12 # MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
13 # GNU General Public License for more details.
14
15 # You should have received a copy of the GNU General Public License
16 # along with this program; if not, write to the Free Software
17 # Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301, USA.
18
19 # http://www.gnu.org/copyleft/gpl.html
20
21 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
22
23 # This program is part of the Biopieces framework (www.biopieces.org).
24
25 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>> DESCRIPTION <<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
26
27 # Swap lower case sequence to uppercase and visa versa for all sequences in the stream.
28
29 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
30
31
32 require 'maasha/biopieces'
33
34 options = Biopieces.options_parse(ARGV, cast = [])
35
36 Biopieces.open(options[:stream_in], options[:stream_out]) do |input, output|
37 input.each_record do |record|
38 record[:SEQ].swapcase! if record.has_key? :SEQ
39 output.puts record
40 end
41 end
42
43
44 # >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>><<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
45
46
47 __END__
Let us go over the different sections in this minimalistic Biopiece:
Line 1. First we have the hashbang line #!/usr/bin/env ruby, note that we use env ruby and not a fixed path!
Lines 3-19. Next we have the license text.
Lines 21-29. This section contains a short description that typically is a copy of the Summary section from the wiki file.
Line 32. Here we have a section with require statements. We always have the require 'biopieces'
line and you are free to require other classes or modules.
Line 34. In this section we can define option casts, which instructs the Biopiece how to handle command line options. These casts are added to the 4 mandatory options that are included in all Biopieces:
{:long => 'help', :short => '?', :type => 'flag', :mandatory => false, :default => nil, :allowed => nil, :disallowed => nil}
{:long => 'stream_in', :short => 'I', :type => 'files!', :mandatory => false, :default => nil, :allowed => nil, :disallowed => nil}
{:long => 'stream_out', :short => 'O', :type => 'file', :mandatory => false, :default => nil, :allowed => nil, :disallowed => nil}
{:long => 'verbose', :short => 'v', :type => 'flag', :mandatory => false, :default => nil, :allowed => nil, :disallowed => nil}
The different keys are:
- :long The long option name - do reuse sensible names from other Biopieces!
- :short The short option name - do reuse sensible names from other Biopieces!
- :type See the type explanations in the [Howto write usage information] section.
- :mandatory Indicate if the option must have an argument.
- :default Allow a default value or a comma seperated string of values depending on the type.
- :allowed Allow a comma seperated string of allowed arguments.
- :disallowed A comma seperated string of arguments not allowed.
Also, the command line options are passed according to the specified casts. If you need more
advanced option checking than provided by biopieces.rb, you will have to do it yourself after
bp.parse, e.g.:
raise "both --database and --genome specified" if options.has_key? "genome" and options.has_key? "database"
raise "no --database or --genome specified" if not options.has_key? "genome" and not options.has_key? "database"
Lines 36-41. Here we have the main iterative loop of the Biopiece demonstrating how to manipulate
records read from input and written to output.
If you need temporary files you can get a temporary directory by calling Biopieces.mktmpdir which
will create a directory in the $BP_TMP directory based on username and session id. The path to this
temporary directory is returned, and the directory is automagically destroyed when the Biopieces
execution is done - also, if the Biopiece is interupted the now stale temporary directory will be
cleaned out when any other Biopiece is executed.
Use the verbose option to allow for messages to show progress or debug:
$stderr.puts "Verbose message" if options.has_key? "verbose"
Finally, add some tests to the Biopieces testing suite to check that everything is working. See Howto write tests for the Biopieces test suite.
To be written ...
To be written ...
The advantage of having a testing suite is that it will alert you to any bugs accidently introduced
in the code during further development of the Biopieces. Basically testing is done by having an
input and a verified result file used for testing a single Biopiece at a time. The input file,
which is typically a stream which is read with the -I switch is read with the Biopieces subject
to testing and the output is written to a temporary file with the -O option. The testing suite
checks if the temporary file and the result file are identical. If they are identical, the test is
OK otherwise it is a FAIL. Biopieces for parsing different types of data will not be reading data
from a stream file, but rather from a sample file with the appropriate data using the -i switch
e.g. a FASTA file in the case of read_fasta. Similary, Biopieces for writing different types of
data will be outputting that data using the -o switch. Sample files are loced in
$BP_DIR/tests/in and result files in $BP_DIR/tests/out while the actual test files are
located in $BP_DIR/tests/test. A minimal structure of the $BP_DIR/tests directory is showed
below:
$BP_DIR/tests/
|-- in
| |-- align_seq.in
| |-- grab.in
| |-- grab.in.pat
| |-- read_fasta.in
| `-- write_fasta.in
|-- lib
| `-- test.sh
|-- out
| |-- align_seq.out.1
| |-- grab.out.1
| |-- grab.out.2
| |-- read_fasta.out.1
| |-- read_fasta.out.2
| |-- write_fasta.out.1
| `-- write_fasta.out.2
|-- test
| |-- test_align_seq
| |-- test_grab
| |-- test_read_fasta
| `-- test_write_fasta
`-- test_all
The tests are written as shell scripts, but are really simple. Lets look at the test for align_seq located here $BP_DIR/tests/test/test_align_seq:
#!/bin/bash
source "$BP_DIR/tests/lib/test.sh"
run "$bp -I $in -O $tmp"
assert_no_diff $tmp $out.1
rm $tmp
This test file contains a shebang line (the one starting with #!) and a source line that make sure
the variables $bp, $in, $out, and $tmp are set. Finnally the test file contains, in this
case, only one test. The test consists of a run command, a assert_no_diff command, and the rm
command. The run command allows us to write a Biopiece command in short hand. In the above
example we will in fact run the command:
align_seq -I /Users/maasha/biopieces/tests/in/align_seq.in -O /Users/maasha/BP_TMP/align_seq.out.1
The $bp variable contains the name of the Biopiece based on the name convention used (the
Biopiece tested is prefixed with test_ like in test_align_seq). The $in variable contains the
path to the input file and the $tmp variable contains the path to a temporary file for outputting
the test result. If we are running multiple tests that give different output we postfix the files
with a forthrunning number.
The assert_no_diff command checks if the resulting $tmp file differs from the validated $out
file, and the result is OK if the files are identical, otherwise the result is FAIL.
Finally, the rm command is used to remove the $tmp file.
Here is an example of the output of the testing suite:
maasha@mel:~$ $BP_DIR/tests/test_all
Testing align_seq -I /Users/maasha/biopieces/tests/in/align_seq.in -O /Users/maasha/BP_TMP/align_seq.out.1 ... OK
Testing grab -I /Users/maasha/biopieces/tests/in/grab.in -p SEQ -O /Users/maasha/BP_TMP/grab.out.1 ... OK
Testing grab -I /Users/maasha/biopieces/tests/in/grab.in -p SEQ,COUNT -O /Users/maasha/BP_TMP/grab.out.2 ... OK
Testing read_fasta -i /Users/maasha/biopieces/tests/in/read_fasta.in -O /Users/maasha/BP_TMP/read_fasta.out.1 ... OK
Testing read_fasta -i /Users/maasha/biopieces/tests/in/read_fasta.in -n 1 -O /Users/maasha/BP_TMP/read_fasta.out.2 ... OK
Testing write_fasta -I /Users/maasha/biopieces/tests/in/write_fasta.in -o /Users/maasha/BP_TMP/write_fasta.out.1 -x ... OK
Testing write_fasta -I /Users/maasha/biopieces/tests/in/write_fasta.in -w 4 -o /Users/maasha/BP_TMP/write_fasta.out.2 -x ... OK
Biopieces tested: 4 Tests run: 7 OK: 7 FAIL: 0 Time: 1 secs
Howto get a Biopiece included at www.biopieces.org
Submit your Biopiece and wiki page to mail@maasha.dk and it will be included after a review.