bamliquidator

Count the number of base pair reads in each bin of each chromosome in the bam file(s) at the given directory, and then normalize, plot, and summarize the counts in the output directory. For additional help, please see https://github.com/BradnerLab/pipeline/wiki
positional arguments: ucsc_chrom_sizes Tab delimited text file with the first column naming the chromosome (e.g. chr1), the third column naming the genome type (e.g. mm8), and the fifth column naming the number of base pairs in the reference chromosome. bam_file_path The directory to recursively search for .bam files for counting. Every .bam file must have a corresponding .bai file at the same location. To count just a single file, provide the .bam file path instead of a directory. The parent directory of each .bam file is interpreted as the cell type (e.g. mm1s might be an appropriate directory name). Bam files in the same directory are grouped together for plotting. Plots use normalized counts, such that all .bam files in the same directory have bin counts that add up to 1 for each chromosome. The .bam file name is also required to contain the genome type so that the corresponding entries in the ucsc_chrom_sizes file can be used. If your .bam files are not in this directory format, please consider creating a directory of sym links to your actual .bam and .bai files. If the .bam file already has 1 or more reads in the HDF5 counts file, then that .bam file is skipped.
optional arguments: -h, --help show this help message and exit --output_directory OUTPUT_DIRECTORY Directory to create and output the h5 and/or html files to (aborts if already exists). Default is "./output". --bin_counts_file BIN_COUNTS_FILE HDF5 counts file from a prior run to be appended to. If unspecified, defaults to creating a new file "bin_counts.h5" in the output directory. --bin_size BIN_SIZE Number of base pairs in each bin -- the smaller the bin size the longer the runtime and the larger the data files (default is 100000).
#### bamliquidator
bamliquidator is run from the command line with required positional arguments:
$ ./bamliquidator [ bamliquidator ] output to stdout
- bam file (.bai file has to be at same location)
- chromosome
- start
- stop
- strand +/-, use dot (.) for both strands
- number of summary points
- extension length
Example counting the number of reads on both strands from base pair 100 to 200 on chromosome 1 (inclusive):
$ bamliquidator 04032013_D1L57ACXX_4.TTAGGC.hg18.bwt.sorted.bam chr1 100 200 . 1 0 120 $
(TODO: add examples with summary points > 1, and explain what extension length does)
<a name="flattener"/>
#### flattener.py
* flattener.py writes hdf5 files to text files and is run from the command line with required positional arguments, e.g.
$ python flattener.py sorted_summary ../output/normalized_counts.h5 ../output $ head ../output/chr1.tab bin_number avg_cell_type_percentile cell_types_gte_95th_percentile cell_types_lt_95th_percentile lines_gte_95th_percentile lines_lt_95th_percentile cell_types_gte_5th_percentile cell_types_lt_5th_percentile lines_gte_5th_percentile lines_lt_5th_percentile 1544 100.0 1 0 1 0 1 0 1 0 916 99.959546925566343 1 0 1 0 1 0 1 0 2295 99.919093851132686 1 0 1 0 1 0 1 0 1654 99.878640776699029 1 0 1 0 1 0 1 0 1211 99.838187702265373 1 0 1 0 1 0 1 0 160 99.797734627831716 1 0 1 0 1 0 1 0 322 99.757281553398059 1 0 1 0 1 0 1 0 168 99.716828478964402 1 0 1 0 1 0 1 0 2263 99.676375404530745 1 0 1 0 1 0 1 0
* flattener.py is in the bamliquidator_internal directory
* flattener.py usage is described with the --help argument:
$ python flattener.py --help usage: flattener.py [-h] table h5_file output_directory
Writes bamliquidator_batch.py hdf5 tables into tab delimited text files, one for each chromosome. Note that this is provided as a convenience, but it is hoped that the hdf5 files will be used directly since they are much more efficient to work with -- e.g. please see http://www.pytables.org/ for easy to use Python APIs and http://www.hdfgroup.org/products/java/hdf-java- html/hdfview/ for an easy to use GUI for browsing HDF5 files. For more info, please see https://github.com/BradnerLab/pipeline/wiki/bamliquidator .
positional arguments: table the table to write to hdf5, e.g. "counts" for bin_counts.h5, or one of the following for normalized_counts.h5: "normalized_counts", "sorted_summary", or "summary" h5_file the hdf5 file generated by bamliquidator_batch.py output_directory directory to store the tab files (must already exist)
optional arguments: -h, --help show this help message and exit
<a name="Performance"/>
# Performance
CPU | Memory | Storage | OS | Liquidation seconds (cold/warmed) | Batch seconds (cold/warmed) | Notes
----|--------|---------|----|-----------------------------------|-----------------------------|------
2GHz Core i7-3667U (dual core) | 8 GB 1600MHz DDR3 | Apple SSD TS128E | Mac OS 10.8.5 | 22.070482 / 20.853430 | 26.841s / 25.203s | git commit [566a2eb](https://github.com/BradnerLab/pipeline/tree/566a2eb86a5a2224780a666d60bef26fc7f66bc2)
2.1GHz Opteron 6272 (32 cores) | 128 GB | ~12 MB/sec NAS | Ubuntu 12.04.4 LTS | 111.846415 / 11.771622 | 118.630 / 18.015 | git commit [566a2eb](https://github.com/BradnerLab/pipeline/tree/566a2eb86a5a2224780a666d60bef26fc7f66bc2)
2.1GHz Opteron 6272 (32 cores) | 128 GB | Dual SSD | Ubuntu 12.04.4 LTS | 11.629656 / 12.311273 | 20.531 / 18.841s | git commit [566a2eb](https://github.com/BradnerLab/pipeline/tree/566a2eb86a5a2224780a666d60bef26fc7f66bc2)
Tests results are from the following command:
time python bamliquidator_batch.py path_foo/ucsc_chromSize.txt path_bar/04032013_D1L57ACXX_4.TTAGGC.hg18.bwt.sorted.bam
The batch time is the real time reported by the time command, and the liquidation time is the time reported in the output formatted like "Liquidation completed in 22.070482 seconds". The first time is from a cold run, and the second time is from a consecutive run which probably utilizes the bam file cached in RAM. To ensure the cold run is really cold, execute with a fresh boot of the computer, or on Linux [clear the cache](http://www.linuxinsight.com/proc_sys_vm_drop_caches.html) by running `sync` followed by `echo 3 > /proc/sys/vm/drop_caches` .
Please email jdimatteo@gmail.com to share your performance results. Please use the same files for testing: the [.bam file](https://www.dropbox.com/s/bu75ojqr2ibkf57/04032013_D1L57ACXX_4.TTAGGC.hg18.bwt.sorted.bam), [.bai file](https://www.dropbox.com/s/a71ngagu2k8pgiv/04032013_D1L57ACXX_4.TTAGGC.hg18.bwt.sorted.bam.bai), and [ucsc_chromSize.txt](https://www.dropbox.com/s/ixwlnlz3mvwx5gn/ucsc_chromSize.txt) files; note that this .bam file is a processed version of a publicly available dataset that can be found at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44931 .
<a name="Developers"/>
# Developers
* bamliquidator was developed by Xin Zhong in the laboratory of Ting Wang at Washington University St.
* bamliquidator_batch.py/bamliquidator_batch were developed by John DiMatteo under the direction of Charles Lin (laboratory of James Bradner, Dana-Farber Cancer Institute)
* additional contributors are welcome, please see [Collaboration Workflow](Collaboration-Workflow)
* source code is available under [The MIT License](http://opensource.org/licenses/MIT): https://github.com/BradnerLab/pipeline
<a name="check-list"/>
#### Developer Getting Started Check List
1. install dependencies: SAMtools, HDF5, boost, C++11 (clang/libc++), tcmalloc, PyTables (version 3 or later), Bokeh, NumPy
* Ubuntu 13.10 or later
* install dependencies under [Install](#Install), then install these:
````
sudo apt-get install git libbam-dev libhdf5-serial-dev libboost-dev libgoogle-perftools-dev clang-3.4 libc++-dev
````
* Ubuntu 12.04 LTS
* dependencies in default apt repo:
````
sudo apt-get install git libbam-dev libhdf5-serial-dev libboost-dev libgoogle-perftools-dev python-numpy python-pandas python-redis python-pip
````
* install clang:
````
sudo apt-get install python-software-properties
sudo add-apt-repository ppa:nmi/llvm-3.3
sudo apt-get update
sudo apt-get install clang-3.3
````
* install libc++
* review instructions at http://libcxx.llvm.org/ , but the following worked for me:
````
jdm@tod:~/Downloads$ svn co http://llvm.org/svn/llvm-project/libcxx/trunk libcxx
jdm@tod:~/Downloads$ mkdir libcxx-build; cd libcxx-build
jdm@tod:~/Downloads/libcxx-build$ CC=clang CXX=clang++ cmake -G "Unix Makefiles" -DLIBCXX_CXX_ABI=libstdc++ -DLIBCXX_LIBSUPCXX_INCLUDE_PATHS="/usr/include/c++/4.6/;/usr/include/c++/4.6/x86_64-linux-gnu/" -DCMAKE_BUILD_TYPE=Release -DCMAKE_INSTALL_PREFIX=/usr/local ../libcxx
jdm@tod:~/Downloads/libcxx-build$ make
jdm@tod:~/Downloads/libcxx-build$ sudo make install
````
4. install bokeh
* `sudo pip install bokeh`
* Mac OS X (10.8 or later)
* install [XCode](https://developer.apple.com/xcode/) (5 or later) and the command line utilities for clang and libc++
* to install the command line utilities, go to the Downloads tab within the Xcode Preferences menu and click "Install" next to the Command Line Tools entry
* install and use [homebrew](http://brew.sh/) for the rest of the dependencies, and then run:
* `brew tap homebrew/science`
* `brew install samtools boost hdf5 google-perftools`
* `ln -s /usr/local/Cellar/samtools/0.1.19/include/bam /usr/local/include/samtools`
* the installed samtools version may vary on your system, e.g. replace 0.1.19 with 0.1.20 or whatever number is installed in your /usr/local/Cellar/samtools/ directory
* this is so that the sam.h can be found in a default search path in the same directory "samtools" as setup by the Ubuntu package libbam-dev
2. checkout, build the code, and verify runs
$ git clone -b hot-spots git@github.com:BradnerLab/pipeline.git # todo: remove hot-spots branch after reintegrated $ cd pipeline/bamliquidator_internal $ make $ ./bamliquidator_batch usage: ./bamliquidator_batch cell_type bin_size ucsc_chrom_size_path bam_file_path hdf5_file
e.g. ./bamliquidator_batch mm1s 100000 /grail/annotations/ucsc_chromSize.txt /ifs/labs/bradner/bam/hg18/mm1s/04032013_D1L57ACXX_4.TTAGGC.hg18.bwt.sorted.bam
note that this application is intended to be run from bamliquidator_batch.py -- see https://github.com/BradnerLab/pipeline/wiki for more information $ ../bamliquidator [ bamliquidator ] output to stdout
- bam file (.bai file has to be at same location)
- chromosome
- start
- stop
- strand +/-, use dot (.) for both strands
- number of summary points
- extension length
$
<a name="components"/>
#### Program Components
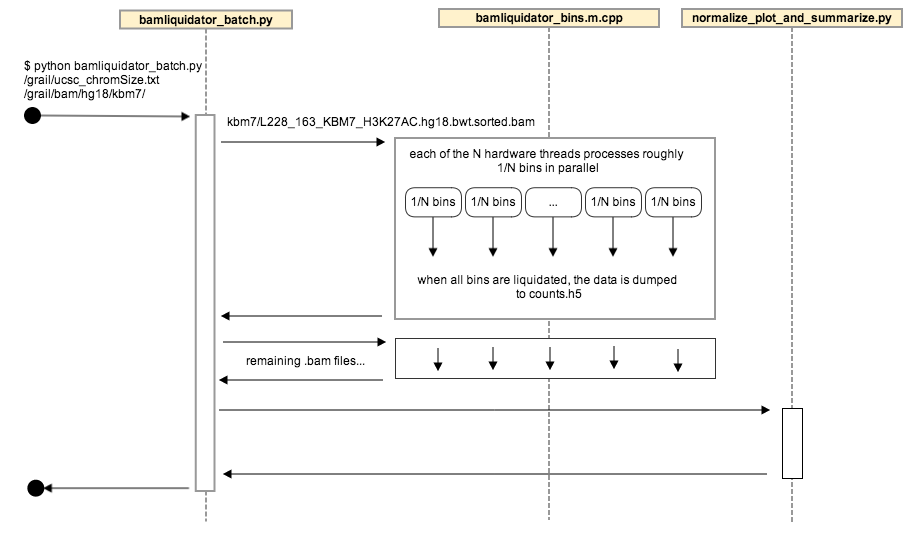
The components of bamliquidator can all be used independently, but are run together by bamliquidator_batch.py:
1. [bamliquidator.h](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidator_internal/bamliquidator.h)/[cpp](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidator_internal/bamliquidator.cpp): generates the raw bin counts
* defines the function `liquidate`, which reads a .bam file to do the counting
* used to create the bamliquidate command line executable ([bamliquidator.m.cpp](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidator_internal/bamliquidator.m.cpp)), and is the core of bamliquidator_batch
2. [bamliquidator_batch.m.cpp](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidator_internal/bamliquidator_batch.m.cpp)
* calls the [liquidate](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidator_internal/bamliquidator.h) function on each chromosome in parallel, and writes the results in HDF5 format
* used to create the bamliquidator_internal/bamliquidator_batch command line utility, which is called by bamliquidator_batch.py
2. [bamliquidator_batch.py](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidatorbatch.py): orchestrates the whole process, and is intended to be the primary user facing application
1. unless an h5 file has been provided for appending to, creates the counts.h5 file in the output directory
2. finds the .bam files to include in processing (see functions all_bam_files_in_directory and bam_files_with_no_counts called by main function)
3. runs bamliquidator_internal/bamliquidator_batch executable on each .bam file (see python function liquidate), storing the results in the counts.h5 file
4. calls the normalize_plot_and_summarize module
3. [normalize_plot_and_summarize.py](https://github.com/BradnerLab/pipeline/blob/hot-spots/bamliquidator_internal/normalize_plot_and_summarize.py): the post-processing of the bin counts
* normalized counts, percentiles, and summaries are calculated and stored in hdf5 tables in the file
"normalized_counts.h5".
* plots are stored in .html files
TODO: update these component links to use master after hot-spots is reintegrated