catalase lab
Catalase is an enzyme that catalyze the breakdown of hydrogen peroxide to water and oxygen.
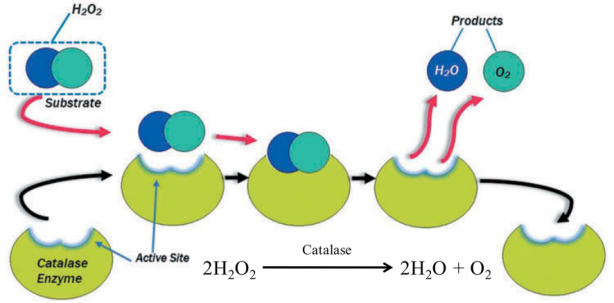
Figure 1
The aim of this practical class is to measure the activity of catalase in permeabilized Saccharomyces cerevisiae yeast cells.
The protocol is based on a combination of a method for preparing permeabilized yeast cells with ethanol described by Trawczyńska & Wójcik 2015 and an assay for peroxide using cobalt ions described by Hadwan 2018.
After the cells have been permeabilized, substrate such as H2O2 can more easily reach the catalase enzyme and the products (H2O and O2) can more easily leave.
The permeabilization protocol is very simple (Figure 2). The yeast cells are incubated with ethanol for 20-30 min in a phosphate buffer.
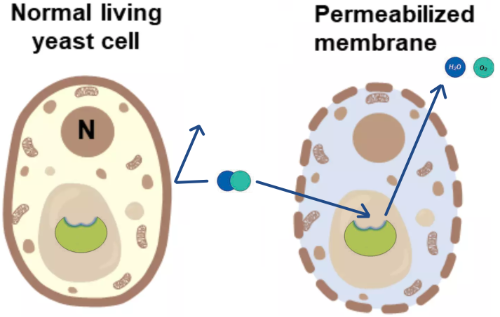
Figure 2
The assay used is an [assay](enzyme activity) where yeast catalase is mixed with hydrogen peroxide for a specific time at 37°C. During this time some of the hydrogen peroxide is broken down to oxygen and water.
After the reaction has stopped, the remainder of the peroxide is measured by allowing the hydrogen peroxide to oxidize cobalt(II) to cobalt(III) which can be measured with a spectrophotometer.
The oxidation is performed in the presence of carbonate ions which causes differently colored complexes with cobalt(II) and cobalt(III) (Figure 3).

Figure 3
Cobalt(III) carbonate (green in Figure 2) has an absorbance peak at 440 nm where Cobalt(II) has very little absorbance.

Figure 4
{kind=link}
Figure 5
All absorbance values are taken with a spectrophotometer calibrated against air without a cuvette (Figure 5).
The Standard reaction in Figure 5 contain hydrogen peroxide and and cobalt(II) carbonate. The peroxide has oxidized cobalt(II) to cobalt(III) (green).
The Test reaction contain H2O2 and cells. Some of the peroxide has been consumed by the catalase in the cells, so the absorbance value at 440 nm should be lower than for the Standard reaction. The Blank contain cobalt(II) carbonate, no peroxide and no cells. The Standard - Blank represent all the green color possible in the assay. Test - Blank represent the green color produced by the leftover H2O2. Therefore, the green color represented by the catalase H2O2 consumption is given by Standard - Blank - (Test - Blank).
There is however one problem left. The cells may also cause absorption by light scattering (Optical Density, OD). We risk a Test reading that is too high, underestimating the real activity. To correct for this, we need a "Cells" cuvette contain only the same number of cells in water or phosphate buffer as in Test. We subtract the OD of the cells (Cells - Water), leading to the final expression (The Blank cancel out and is not needed):
{kind=link}
Figure 6
At this point, we have absorbance change or ∆A per 10 min. What we want is µmol / min which is the definition of the classical unit.
We use a standard curve (Figure 7) to calculate the actual concentration of the samples. The activity is then calculated from the concentration difference between the Standard and the sample divided by the reaction time.
A 10 mM hydrogen peroxide solution was serially diluted (Figure 7) by preparing twelve Eppendorf tubes with 300 µL of ddH2O. One milliliter hydrogen peroxide was transferred to the first tube, the contents were mixed by pipetting up and down 5-6 times followed by transferring 1 mL to the next tube. This means that the last tube (12) eventually had a final volume of 1.3 ,L while all preceding tubes had 300 µL.
The hydrogen peroxide concentration in the first tube is (1 mL * 10 mM) / 1.3 mL = 7.6923 mM. The dilution factor is the same for all tubes 1/1.3 so the concentration of each tube can be calculated using the formula
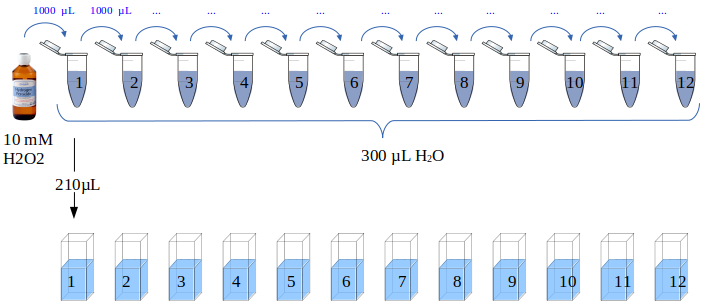
Figure 7
Cuvettes with 210 µL of each dilution were prepared, twelve in total. Working solution (800 µL) (Series#1 and #2 below) was added to each cuvette followed by 10 min incubation at room temperature under darkness. Two replicates were prepared and the absorbance at 440 nm was measured with a GENESYS20 spectrophotometer.


The absorbance readings were collected in the table below.
| Tube | Concentration (mM) | Series#1 | Series#2 |
|---|---|---|---|
| 1 | 7.692 | 0.450 | 0.414 |
| 2 | 5.917 | 0.394 | 0.361 |
| 3 | 4.551 | 0.345 | 0.307 |
| 4 | 3.501 | 0.291 | 0.263 |
| 5 | 2.693 | 0.248 | 0.225 |
| 6 | 2.072 | 0.213 | 0.194 |
| 7 | 1.594 | 0.197 | 0.166 |
| 8 | 1.226 | 0.160 | 0.148 |
| 9 | 0.943 | 0.143 | 0.134 |
| 10 | 0.725 | 0.123 | 0.115 |
| 11 | 0.558 | 0.109 | 0.113 |
| 12 | 0.429 | 0.113 | 0.095 |
The average absorbance was plotted against the hydrogen peroxide concentration resulting in a reasonably linear relationship (Figure 8).
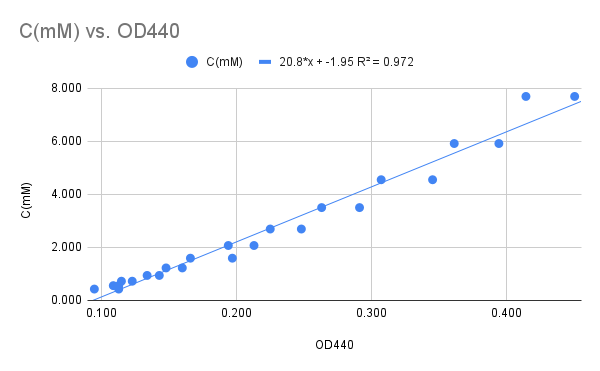
Figure 8
The graph in Figure 8 leads to the linear relationship between Concentration (mM) and A440:
Inoculate two 50 mL glass tubes with ~5 mL of SD medium and incubate at 30 °C for ~48h.
- Tube#1 CENPK111-32D + pTA1
- Tube#2 CENPK111-32D + pTA1_TDH3_ScCTT1_PGI1
At 10:00 Inoculate two 100 mL erlenmeyers with the same medium using 1 mL of the two pre-cultures.
Put both cultures on ice. Measure the optical density (OD) at 600 nm. Add fresh medium to the culture with the higher OD in order to set the two cultures to the same optical density.
Keep the cultures on ice until used.
| Group | Strain | Plasmid vector |
|---|---|---|
| 1 | CENPK111-32D | pTA1 (empty) |
| 2 | " | " |
| 3 | " | pTA1_TDH3_ScCTT1_PGI1 |
| 4 | " | " |
- Each group (1-4) should take out 1 mL of the culture indicated in the table above in a an empty 1.5 mL Eppendorf tube.
- Spin down the culture for 30 s in a microcentrifuge.
- Remove supernatant with a P1000 pipette.
- Add 1 mL phosphate buffer.
- Resuspend cells by pipetting up&down with the same tip.
- Spin down culture for 30 s.
- Remove supernatant.
- Add 500 µL phosphate buffer.
- Resuspend cells.
- Add 500 µL ethanol.
- Mix the content of the tube by inverting the tube 2-3 times.
- Incubate for 20 min @ room temperature on the bench During the incubation, proceed with the protocol below
- Mark five empty plastic cuvettes with a marker, T1 and T2 for Tests, S for standard, C for cells and W for water.
- Pour ~1 mL of hydrogen peroxide into a 1.5 mL Eppendorf tube and store on your bench.
- Pour ~5 mL of working solution into a 15 mL FALCON tube and store on your bench.
Wait until the permeabilized cells are ready.
-
Dilution 1:10 Add 900 µL of distilled water to an empty Eppendorf tube. Add 100 µL of the permeabilized cells. Mix the content of the tubes well before and after.
-
Add 940 µL of distilled water to a the "C" cuvette. Add 70 µL diluted cells to the cuvette. and measure the absorbance for each cuvette at 440 nm against air.
-
Add the components indicated to the bottom of the cuvette. Prepare the cuvettes in the indicated order:
Standard cuvette
- 70 µL distilled water
- 140 µL hydrogen peroxide
Test 1 cuvette
- 70 µL diluted cells
- 140 µL hydrogen peroxide
Test 2 cuvette
- 35 µL diluted cells
- 35 µL distilled water
- 140 µL hydrogen peroxide
Incubate cuvettes at 37 °C in a water bath for 10 min. This timing is critical! Add 800 µL working solution to each cuvette (except the C cuvette). Mix by inversion with a piece of parafilm if necessary. Incubate at room temperature for 10 min in the dark. You can put the cuvettes in one of the empty drawers in the lab.
Record the absorbance for each cuvette at 440 nm against air. Take a picture of the cuvettes with your phone.
Add the data to the Google Spreadsheet.
This practical class was based on a combination of methods described in the two publications below:
Trawczyńska, I., & Wójcik, M. (2015). Optimization of permeabilization process of yeast cells for catalase activity using response surface methodology. Biotechnology, Biotechnological Equipment, 29(1), 72–77. link
Hadwan, M. H. (2018). Simple spectrophotometric assay for measuring catalase activity in biological tissues. BMC Biochemistry, 19(1), 7. link
Kaplan, J. G. (1963). THE REVERSION OF CATALASE DURING GROWTH OF YEAST IN ANAEROBIOSIS. The Journal of General Physiology, 47, 103–115. link
Martins, D., & English, A. M. (2014). Catalase activity is stimulated by H(2)O(2) in rich culture medium and is required for H(2)O(2) resistance and adaptation in yeast. Redox Biology, 2, 308–313. link
This buffer can be prepared by by mixing solutions A and B at a ratio of 1:1.5. Solution A: 6.81 g/L of KH2PO4 Solution B: 8.90 g/L Na2HPO4 in ultrapure water
25 mL Sodium bicarbonate (NaHCO3) solution
Prepared by dissolving 2 g (MW 84.01 g/mol) in 25 ml ultrapure water. This solution should be stored at room temperature.
Prepared by dissolving 0.51 g of Co(NO3)2 x 6H2O in 25 ml of distilled water. This solution should be stored at room temperature in the dark.
Prepared by dissolving 0.25 g of (NaPO3)6 in 25 ml of distilled water. This solution should be stored at room temperature.
Prepared by adding 12 µL of 30% hydrogen peroxide to 10 ml of 50 mM phosphate buffer (pH 7.0). This solution should be freshly prepared before class.
This solution is made by mixing cobalt (II) solution, Graham salt solution and Sodium bicarbonate solution. Prepared by mixing 5 ml of cobalt (II) solution, 5 ml of Graham salt solution, and 90 ml of sodium bicarbonate solution. The order in which these substances are added is very important for obtaining accurate results. This solution should be stored at room temperature in the dark.
- 6.7 g/L Yeast Nitrogen Base (YNB) WITHOUT AMINO ACIDS
- 20 g/L glucose This is a liquid medium.
- Six 100 mL empty Erlenmeyer flasks with cotton stoppers (sterile)
- Eight empty new FALCON tubes 50 mL
- Eppendorf tubes 1.5 mL (sterile or not)
- Blue tips
- Yellow tips
- Cuvettes
- 37 °C water bath
- boat for cuvettes
- microcentrifuge
- vortex
- P1000 pipettes
- P200 pipettes
🇵🇹
Material & Reagentes:
(6-feria) 6 x 100 mL Erlenmeyers com algodão, estereis
500 mL meio SD (Synthetic Defined) - 6.7 g/L Yeast Nitrogen Base (YNB) WITHOUT AMINO ACIDS - 20 g/L glicose
Dia da aula:
Oito tubos FALCON tubes 50 mL novos pipetas P1000, P200 Pontas amarelas, azuis vortex microcentrifuga banho maria 37 °C (barco para cuvettes) Cuvettes Eppendorfs 1.5 mL
250 mL Tampão de fosfato (pH 7,0, 50 mM) numa proporção de 1: 1.5 (a:b):
(a) 6.8 g/L de KH2PO4 em água destilada
(b) 8.90 g/L de Na2HPO4em água destilada
25 mL Solução cobalto (II): 10,15 g de Co(NO3)2 · 6H2O em 500 mL de água destilada
25 mL Solução de sal de Graham (hexametafosfato de sódio): 5 g (NaPO3)6 em 500 mL de água destilada
25 mL Solução de bicarbonato de sódio: 2 g (MW 84.01 g/mol) em 25 ml agua ultrapura
Solução de trabalho: 5 mL de solução de cobalto (II), 5 mL de solução de sal de Graham, e 90 mL de solução de bicarbonato de sódio. Feito no dia antes da aula.
Peróxido de hidrogénio (10 mM): Adicionar 12 µL L de peróxido de hidrogénio (30%) para um volume final de 10 mL de tampão fosfato (pH 7,0, 50 mM).
Etanol 95-99%: 50 mL