The root mean Square Deviation (RMSD) is the most common metric for measuring structural similarity between two structures. It is typically used in molecular biology, chemistry, and computational chemistry.
However, the result can become misleadingly large unless the input data has pre-optimized translation and rotation of the molecules in question. This solution will perform this optimization before calculating optimal (minimal) RMSD values.
Additionally, if the atoms in the molecules are not correctly ordered, optimal rotation is impossible to achieve. This tool utilizes several ways to solve this problem.
For more details, see below and read RMSD and Kabsch algorithm.
Overview
The easiest is to get the program via pip.
pip install rmsdThere is only one Python file, so you can also download calculate_rmsd.py and put it in your bin folder.
wget -O calculate_rmsd https://raw.githubusercontent.com/charnley/rmsd/master/rmsd/calculate_rmsd.py
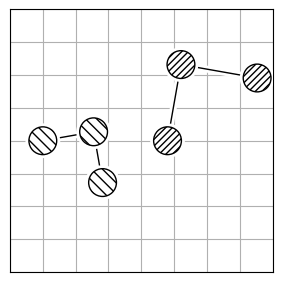
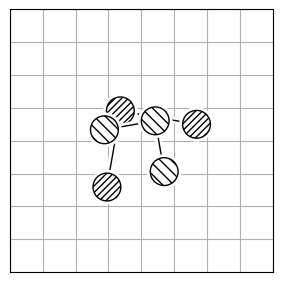
chmod +x calculate_rmsdTo calculate the structural difference between two molecules, you might initially compute the RMSD directly (Figure 1.a). However, this straightforward approach could give you a misleadingly large value. To get the true minimal RMSD, you must adjust for translation (Figure 1.b) and rotation (Figure 1.c). This process aligns the two molecules best, ensuring the RMSD accurately reflects their structural similarity after optimal alignment.
| 1.a | 1.b | 1.c |
|---|---|---|
 |
 |
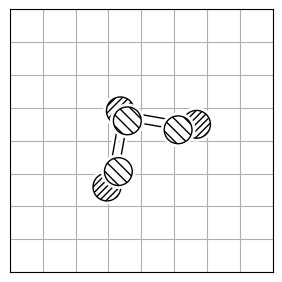 |
| RMSD = 2.8 | RMSD = 0.8 | RMSD = 0.2 |
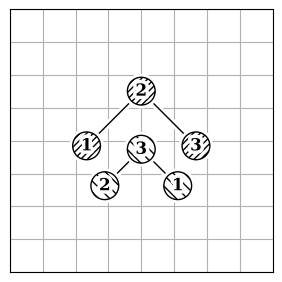
Atom reordering methods are used when the atoms in two molecules are not in the same order (Figure 2.a).
While brute-force through all possible atom combinations and calculating the optimal rotation for each is possible, this approach is computationally infeasible for large structures, as it scales
Each method has limitations because finding the best atom mapping depends on properly aligning structures. This is usually done by comparing atom-pair distances. If the molecules are aligned, using the Hungarian cost minimization for atom distance works well. If not, you can align the Inertia eigenvectors (Figure 2.b) as an approximation to align the molecules. Or, use atomic descriptors (Figure 2.c), independent of the coordinate system, to reorder the atoms. Note that all reordering methods have limitations and drawbacks, and the actual order might not be found.
| 2.a | 2.b | 2.c |
|---|---|---|
 |
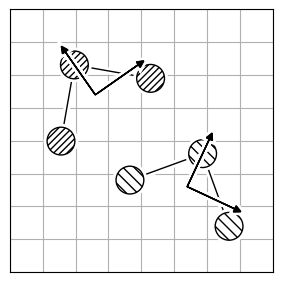 |
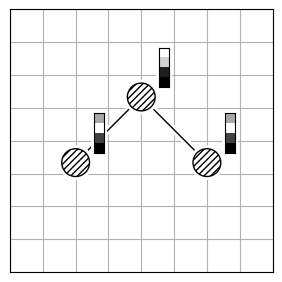 |
Use calculate_rmsd --help to see all the features. Usage is pretty straight
forward, call calculate_rmsd with two structures in either .xyz or
.pdb. In this example, Ethane has the same structure but is
translated in space, so the RMSD should be zero.
calculate_rmsd tests/ethane.xyz tests/ethane_translate.xyzIt is also possible to ignore all hydrogens (useful for larger molecules where hydrogens move around indistinguishable) and print the rotated structure for visual comparison. The output will be in XYZ format.
calculate_rmsd --no-hydrogen --print tests/ethane.xyz tests/ethane_mini.xyzIf the atoms are scrambled and not aligned, you can use the --reorder
argument, which will align the atoms from structure B onto A.
Use --reorder-method to select the reordering method.
Choose between
Inertia aligned Hungarian distance inertia-hungarian (default),
Hungarian distance hungarian (if the structure is already aligned),
sorted distance distance,
atomic representation qml,
and brute force brute (for reference, don't use this).
More details on which to use in --help.
calculate_rmsd --reorder tests/water_16.xyz tests/water_16_idx.xyzIf you want to run multiple calculations simultaneously, it's best not to rely solely on the script.
Instead, you can use GNU Parallel to handle this efficiently. For example, compare all ethane_* molecules using two cores and print one file and the RMSD per line.
Bash is good for stuff like that.
find tests/resources -name "ethane_*xyz" | parallel -j2 "echo -n '{} ' && calculate_rmsd --reorder --no-hydrogen tests/resources/ethane.xyz {}"It is also possible to use RMSD as a library in other scripts; see tests/* for example usage.
Found a bug? Submit issues or pull requests on GitHub.
Note on PDB format. Protein Data Bank format (PDB) is column-based; however, countless examples of non-standard .pdb files exist.
We try to read them, but if you have trouble reading the file, check if the file format is compliant with PDB.
For example, some hydrogens are noted as HG11, which we assume is not mercury.
Please cite this project when using it for scientific publications. And cite the relevant methods implemented.
Implementation: Calculate Root-mean-square deviation (RMSD) of Two Molecules Using Rotation, GitHub, http://github.com/charnley/rmsd, <git commit hash or version number>
| Method | Argument | Citation |
|---|---|---|
| Kabsch | --rotation-method kabsch (Default) |
Wolfgang Kabsch (1976), Acta Crystallographica, A32:922-923 |
| Quaternion | --rotation-method quaternion |
Walker, Shao & Volz (1991), CVGIP: Image Understanding, 54:358-367, |
| Distance Hungarian Assignment | --reorder-method inertia-hungarian (Default) |
Crouse (2016). Vol. 52, Issue 4, pp. 1679–1696, IEEE. |
| FCHL19 | --reorder-method qml |
Christensen et al (2020), J. Chem. Phys. 152, 044107 |