- 실행결과

- NGS(Next-Generation Sequencing) 기술이 발전하며 유전정보 해석에 필요한 시간과 노력이 많이 감소되었다. 이와 더불어 최적의 sequence alignment를 찾는 것의 중요성 또한 강조되고 있다.
- Sequence alignments분석의 목적은 관심 있는 서열의 유사성과 차이점을 분석하여 염기와 아미노산 수준에서 서열간의 구조적, 기능적 및 진화론적 관련성을 추론하는 것에 있다.
- 본 프로젝트에서 알고리즘의 구현은 DNA와 Protein 두 가지로 나누어 진행되며, 사용자로 하여금 원하는 alignmet를 선택하여 결과를 확인할 수 있도록 한다.
- Dynamic programming을 통해 임의의 두 sequence를 입력으로 넣으면 최적의 alignment를 찾아내는 알고리즘을 구현하고, 사용자로 하여금 결과와 사용된 알고리즘 정보를 같이 표현해 주는 등 직관적인 인터페이스를 만드는 것을 목적으로 한다.
- 구현한 알고리즘을 바탕으로 공통된 유전자 부분이 병변이나 형질에 영향을 주는 경우가 있는지를 조사하여, 이 또한 함께 출력해 진화론적 관련성 또한 추론해 볼 수 있도록 한다.
- Sequence alignment 관련 methods, models, concepts, and strategies (서적과 논문 활용) http://delab.yonsei.ac.kr/assets/files/publication/legacy/Graph Sequence Alignment Using a Distributed System.pdf http://www.koreascience.kr/article/JAKO200508824135995.kr > http://multalin.toulouse.inra.fr/multalin/multalin.html (Alignment program + Macvector, Vector NTI, clustalx)
- Matrix and Algorithm-Relationship between BLOSUM & PAM
- BLOSUM과 PAM 두 matrices 모두 substitution이라는 전제하에 비교하려는 sequences의 similarity를 보고자 하는 matrix 이다
- Dynamic programming을 통해 sequence alignment를 구현한다. 두 임의의 sequence가 입력으로 들어가면 여러 substitution matrix를 사용하여 자동으로 결과를 비교하고, 최적의 모델을 통해 산출된 score와 gap score, 그리고 alignment 결과를 출력한다.
- 최종적인 결과물을 웹 프로그래밍을 통해 웹에서 사용자들이 편하게 이용할 수 있도록 인터페이스를 구축한다.
- Dynamic programming을 통한 sequence alignment 구현
- 두 가지 방식으로 나누어 구현 → DNA, Protein (*모든 코드는 구글 코랩을 기준으로 작성)
- 두 임의의 sequence가 입력으로 들어가면 설정한 gap score, match score, mismatch score에 맞추어서 최적의 모델을 통해 산출된 score_matrix와 alignment 결과가 출력된다.
- 두 임의의 sequence가 입력으로 들어가면 여러 substitution matrix를 사용하여 자동으로 결과를 비교하고, 최적의 모델을 통해 산출된 score와 alignment 결과를 출력한다.
- Evaluation method: 이미Sequence분석 결과가 나와있는 서열들을 직접 구현한 코드에 넣어보고 최적의 결과를 찾아내는지 확인한다.
- 구현한 알고리즘은 최적의 alignment 결과를 출력해주며, Protein의 경우 8가지 다른 matrix중에서 가장 좋은 alignment를 선택하여 보여준다.
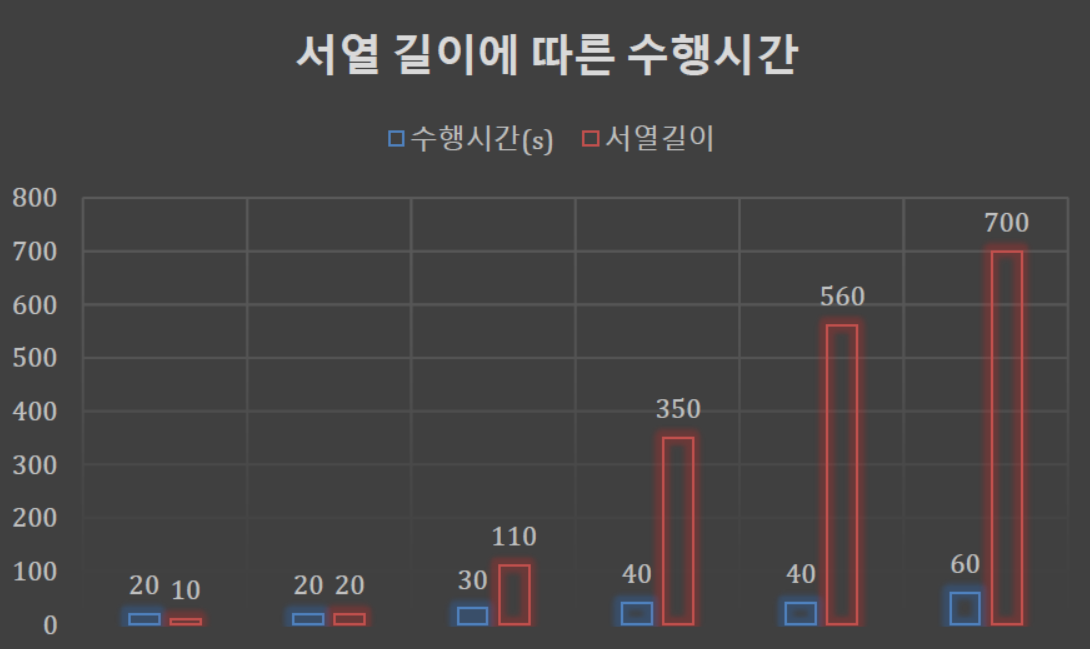
- 알고리즘은 이중 for문을 사용하였기 때문에 O(n^2)의 수행시간을 가진다.
- 따라서 입력의 크기가 증가할수록 수행시간도 증가한다.
- 두 서열을 aligning하는 과정은 database similarity searching과 multiple sequence alignment의 기초가 된다.
- 고안한 알고리즘은 특정 scoring function이 주어졌을 때 최적의 결과를 보장한다.
- 바이오인포메틱스에서 서열 정렬은 DNA, RNA or Protein의 서열을 정렬하는 방법으로 유사성을 지닌 지역을 확인하는 과정이며, 이러한 유사성은 기능, 구조, 진화적인 관계에 의한 결과라고 볼 수 있다.
- O(n^2) 수행시간을 가지는 알고리즘의 경우 입력이 매우 커지면 수행시간도 매우 커지게 된다. 따라서 수행시간을 줄일 수 있는 방향으로 알고리즘을 개선하는 것이 향후 과제가 될 것이다.