Calulate coverage
By a request, I've made a small guide how to calculate the coverage of aligned reads to some reference
###This method will assume that you have you alignment in a sorted bam file (ofc you can pass a .sam through samtools view)
To call the function you need you can either call bedtools:
bedtools genomecov
or call the sub-program directly:
genomeCoverageBed
To get the coverage for each base (including the bases that are covered by no reads) you simply write:
genomeCoverageBed -ibam youralignment.bam -d
Which will write to STDOUT <HEADER/NAME> <POS> <COVERAGE>
Bedtools will simply report everything in the bamfile in one column. But, you can use samtools to pass the specific question your interested in. For example, you could give a specific name, filter by mapping quality or filter by flags etc.
To use samtools to get the coverage of a specific name in your bam file, say "Chr4" you write:
samtools view -u youralignment.bam Chr4 | genomeCoverageBed -ibam stdin -d
#Yes you write out "stdin"
The functionality of the other flags than -d check this image:
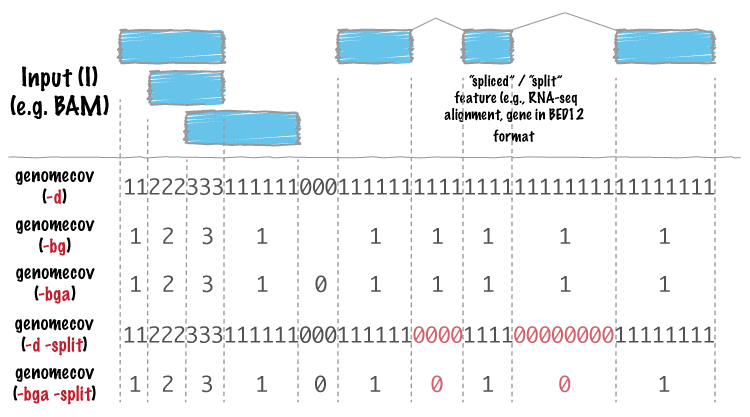
This part will assume you have your alignment in a sorted bam file that is also indexed using samtools!
Ok, so say that you want to do this repeatedly, but you are not happy enough with writing a shell script that does this with bedtools, you could instead use the module pysam in python. I've written a CNV-calling program that is implementing this (check the CNV_pipe repo to see the python code).
You first need to install pysam (installed on dna.lundberg.gu.se):
pip install pysam
Then you can write the following code:
import pysam
#First import the bam file
bamfile = pysam.AlignmentFile("path/to/my/bamfile.bam", "rb")
# You can now iterate over the bamfile object as:
for pileup in bamfile.pileup("Chr4", 10000, 10020, truncate=True):
print pileup.n
#This will print out the coverage of the position 10000-10020 in the name "Chr4"
#If you want entire Chr4 you simply write:
for pileup in bamfile.pileup("Chr4"):
print pileup.n
# The position of each base can be called with pileup.pos
# If you want to iterate over the entire bamfile a good thing could be to get a tuple with all headers:
headers = bamfile.references
# Or you can get a dictionary including also the length of each name:
header_dict = bamfile.headerPysam will not - as bedtools so kindly do - report the bases with zero coverage by default. But we can circumvent this by a few lines extra :)
import pysam
Y = []
bamfile = pysam.AlignmentFile("path/to/my/bamfile.bam", "rb")
start_pos = 0
stop_pos = 101
for pile in bamfile.pileup("Chr4", start_pos , stop_pos, truncate=True):
while pile.pos != start_pos:
Y.append(0)
start_pos = start_pos + 1
Y.append(pile.n)
start_pos = start_pos + 1
print Y
# Which will print a list with the coverage for the first 100 bases