Mutation-Simulator is a tool for simulating SNPs and SVs in any reference genome with cohesive documentation about the implemented mutations. With Mutation-Simulator, the new file format Random Mutation Tables (RMT) is introduced, which gives more simulation power to the user by creating an interface for highly customized simulations. This can potentially achieve more natural simulation patterns tailored to the reference genome. Mutation-Simulator provides 3 different modes to simulate SNPs, insertions, deletions, tandem duplications, inversions, translocations and interchromosomal translocations from the command line or with highly configurable RMT files.
- The sum of all mutation rates can no longer exceed 0.5
- Major rewrite resulting in performance and code readability gains
- Python version 3.10.0 or higher is now required
- Mutation type length options/keywords have entirely been replaced by minimum and maximum length counterparts
- Standard output path is now the current working directory
_msand_ms_itwill now always be added to the output filenames- Output Fasta file extensions will now be deduced from the input Fasta
-w/--ignore-warningsis now a top level option available in all modes
The simulation of mutations is a useful tool when benchmarking bioinformatics programs for variant identification and read alignment. While other simulation programs provide a set of tools to simulate some types of SNPs or SVs, they are often either lacking features or limited to a specific genome type. The need for a "Swiss Army Knife" of simulation software combined with the wish to define mutation rate hot and cold spots, led towards the development of Mutation-Simulator. This tool is not designed to be used as an evolution or inheritance simulator. It is mainly designed to assist other tools, for example variant callers or read aligners as a benchmark. Mutation-Simulator provides the functionality to edit reference sequences using user specified configuration files (RMT) or command line options. With Mutation-Simulator it is possible to test whether other tools handle SNPs and SVs correctly. RMT files aim to assist the user in creating a benchmarking dataset that is as close to biological data as desired.
- Many common SV types and SNPs supported
- Not restricted to specific genomes / genome types
- Highly configurable
- New RMT file format allows adjusting mutation-rates easily on a base resolution for more realistic mutation patterns
- Regions on the reference can be blocked from mutating (for example centromeres)
- Operates with full genomes or small DNA fragments in Fasta files
- Simple syntax
- VCF documentation of introduced SNPs and Structural Variations (SVs).
- BEDPE documentation of interchromosomal translocations
- SNP simulation supports transitions / transversion rates
- Translocations are marked with INS:ME and DEL:ME in the VCF file
- (Recommended) Create and activate a new Python >= 3.10 environment with pyenv and pyenv-virtualenv.
pyenv install 3.10.0
pyenv virtualenv 3.10.0 mutation-simulator
pyenv activate mutation-simulator- Or Conda
conda create -n py310 python=3.10
conda activate py310- Install with pip
pip install Mutation-Simulator- Python >= 3.10.0
- numpy
- pyfaidx
- tqdm
Mutation-Simulator is executable in 3 modes: Command-line Arguments (ARGS),
Interchromosomal Translocations (IT) and Random Mutation Table (RMT). These
modes can be selected with the second positional argument mode:
mutation-simulator [options] file {mode} ...ARGS represents a quick way to simulate any kind of SV or SNP to a given rate and length uniformly distributed across the reference genome. It provides no option to select specific areas on the genome for the mutation rates to deviate from. All positions will be randomly generated across the genome on all sequences within the reference. The use case of ARGS confines to enabling the user to create a variant of the reference genome if no fine-grained settings need to be considered and solely exists as a quality of life feature.
IT serves the same use case as ARGS, though IT is used to simulate interchromosomal translocations at a specified rate. First two sequences will be randomly paired. Secondly the amount of breakpoints is determined by the specified rate. Each breakpoint is randomly generated independently from the breakpoints of the paired sequence. Breakpoints can occur within the first and last position of the sequences with a minimum distance of one. The sequences after each breakpoint will then be swapped between the paired chromosomes. As well as ARGS, IT does not provide an option to define a different rate for each chromosome in the reference.
RMT is a combination of the aforementioned features with the addition to define ranges of higher or lower mutation rates on each sequence using an RMT file. This file format allows the user to create specific recreatable patterns in which mutations can be generated at given rates and lengths across specific sequences. RMT also features meta-information about the genome, specific IT rates for each sequence in the reference, configurable standard values for unspecified areas, and blocking positions from mutating entirely.
| Option | Option | Description | Default |
|---|---|---|---|
| -sn | -–snp | Rate of SNPs | 0 |
| -snb | -–snpblock | Amount of bases blocked after SNP | 1 |
| -titv | --transitiontransversion | Ratio of transitions / transversions | 1 |
| -in | -–insert | Rate of insertions | 0 |
| -inmin | -–insertminlength | Minimum length of inserts | 1 |
| -inmax | -–insertmaxlength | Maximum length of inserts | 2 |
| -inb | -–insertblock | Amount of bases blocked after insert | 1 |
| -de | -–deletion | Rate of deletions | 0 |
| -demin | -–deletionminlength | Minimum length of deletions | 1 |
| -demax | -–deletionmaxlength | Maximum length of deletions | 2 |
| -deb | -–deletionblock | Amount of bases blocked after deletion | 1 |
| -iv | -–inversion | Rate of inversions | 0 |
| -ivmin | -–inversionminlength | Minimum length of inversion | 2 |
| -ivmax | -–inversionmaxlength | Maximum length of inversion | 3 |
| -ivb | -–inversionblock | Amount of bases blocked after inversion | 1 |
| -du | -–duplication | Rate of duplications | 0 |
| -dumin | -–duplicationminlength | Minimum length of duplications | 1 |
| -dumax | -–duplicationmaxlength | Maximum length of duplications | 2 |
| -dub | -–duplicationblock | Amount of bases blocked after duplication | 1 |
| -tl | -–translocation | Rate of translocations | 0 |
| -tlmin | -–translocationminlength | Minimum length of translocations | 1 |
| -tlmax | -–translocationmaxlength | Maximum length of translocations | 2 |
| -tlb | -–translocationblock | Amount of bases blocked after translocations | 1 |
| -a | -–assembly | Assembly name for the VCF file | "Unknown" |
| -s | -–species | Species name for the VCF file | "Unknown" |
| -n | -–sample | Sample name for the VCF file | "Unknown" |
The table shows all possible command line options in the ARGS mode. All options can be combined as desired. For example:
mutation-simulator file args -sn 0.01 -in 0.005 -inmin 1 -inmax 5Mutation-Simulator will generate the simulated genome named after the reference
file $_ms.fa or $_ms.fasta in the current working directory, together with
the corresponding VCF file $_ms.vcf. This can be changed with the -o or
--output option available across all modes. A Fasta index file for the
reference will be generated automatically during execution.
IT mode only features one additional argument:
mutation-simulator file it interchromosomalrateMutation-Simulator will generate the simulated genome named after the reference
file $_ms_it.fa or $_ms_it.fasta in the current working directory, together
with the corresponding BEDPE file $_ms.bedpe. This can be changed with the
-o or --output option available across all modes. A Fasta index file for
the reference will be generated automatically during execution.
RMT solely requires an RMT file to operate:
mutation-simulator file rmt rmtfileA RMT file contains all the information needed for Mutation-Simulator to generate a highly customized simulation of mutational patterns within a specific genome.
To learn more about RMT files read the RMT Docs. To learn how to create RMT files read the Workflows section.
For convenience, we created some ready-to-use example RMT files for commonly used organisms where all genes are blocked from mutating. These files can be found here.
| Species | RMT | Source GFF |
|---|---|---|
| Arabidopsis thaliana | Arabidopsis_thaliana.rmt | TAIR10_GFF3_genes.gff |
| Danio rerio | Danio_rerio.rmt | Danio_rerio.GRCz11.100.gff3.gz |
| Drosophila melanogaster | Drosophila_melanogaster.rmt | Drosophila_melanogaster.BDGP6.28.100.gff3.gz |
| Homo sapiens | Homo_sapiens.rmt | gencode.v34.annotation.gff3.gz |
| Mus musculus | Mus_musculus.rmt | Mus_musculus.GRCm38.100.gff3.gz |
To simulate SNPs uniformly across all sequences, just pass the Fasta file path and a SNP mutation rate to Mutation-Simulator in args mode.
mutation-simulator genome.fasta args -sn 0.05This command will generate SNPs about every 20th base within the sequences of
the genome.fasta file. The mutated sequences will be saved in
genome_ms.fasta. A transition/ transversion (ti/tv) ratio can be set by the
-titv option. The following example shows a ti/tv ratio of 2:4.
mutation-simulator genome.fasta args -sn 0.05 -titv 0.5If a specific minimum distance between SNPs is desired, the command can be extended by the SNP block option, which specifies a number of blocked bases after each SNP.
mutation-simulator genome.fasta args -sn 0.05 -snb 10 -titv 0.5Note: Mutation-Simulator will always block at least 1 base between mutations.
If a uniform distribution of SVs across the genome is sufficient enough, the same procedure as above can be used. All options of the ARGS mode can be combined in a single command as desired. For example:
mutation-simulator genome.fasta args -sn 0.01 -in 0.01 -de 0.01 -du 0.01 -iv 0.01 -tl 0.01 -inmin 10 -inmax 100The RMT mode is preferred if any specific settings are needed eg.:
- Blocking specific chromosomes from mutating
- Setting hot / cold spots
- Adjusting mutation types, rates or lengths on a per base resolution
To learn more about RMT files read the RMT Docs. To learn how to create RMT files read the Workflows section.
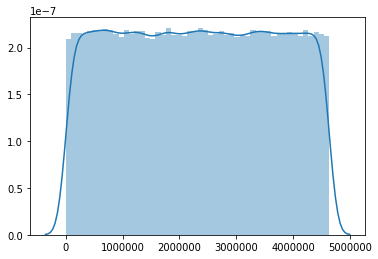
It might be helpful to visualize the mutation distribution of a VCF file, for example to compare the distributions of the VCF file used to generate the RMT file, and the VCF file resulting from running Mutation-Simulator with that RMT file. This can easily be done using bcftools and python:
- First, bgzip and index the VCF file.
bgzip myVariants.vcf; bcftools index myVariants.vcf.gz- Then extract positions and plot the distribution.
# Plotting the mutation distribution for 'chr1'
from pysam import VariantFile
import seaborn as sns
vcf_in = VariantFile("myVariants.vcf.gz")
x = []
for rec in vcf_in.fetch("chr1"):
x.append(rec.pos)
sns.distplot(x)Although, RMT files can be written manually, if data about mutation rate distributions of mutation types along a genomic sequence are available in a suitable format like VCF or GTF/GFF, an RMT file can be created automatically (see Example RMT files). In short, this is done by counting the occurrences of a mutation type in each bin of a user-defined length along the sequence. The rate of the mutation type in that bin is then calculated by dividing the number of occurrences by the length of the interval/bin. This can easily be done in every scripting language, or with a number of available CLI tools.
Of course, the way the VCF or GTF/GFF file was generated strongly influences the represented rates. For example, the VCF file of one sample from a single resequencing project contains the variants between this sample and the reference sequence. The resulting RMT file after VCF to RMT conversion can be used to generate sequences that have the same rate and distribution of variants and thus genetic distance as the resequenced sample. If calculating the rates using all samples from a multi-sample VCF instead, this will result in a sequence with variant rates and distribution of the whole population. Thus, choosing an input file from the desired experimental setup is crucial.
The most simple case is to create an RMT file for a sequence from a sorted VCF file containing one sample and one type of mutation. This example workflow uses the Unix command line, bcftools, bedops and samtools.
- Index the sequence.
samtools faidx sequence.fa- Create a bed file, containing start and end positions of each sequence in the reference.
cat sequence.fa.fai | awk '{ print $1, "0", $2 }' > sequence.bed- Create a bed file of genomic windows, in which the mutations will be counted. Here, the size of the windows can be changed to be more fine-grained or coarse.
bedops --chop 10000 sequence.bed > sequence.10kbwindows.bed- Convert the VCF file to bed format.
cat mutations.vcf | vcf2bed > mutations.bed- Count SNPs across windows with an overlap of 5 kb on both sides. This command determines mutation counts by a sliding-window with 20 kb window size in 10 kb steps.
bedmap --echo --count --range 5000 sequence.10kbwindows.bed mutations.bed > mutations.windows.counts.bedThe resulting mutations.windows.counts.bed file will look like this:
Chr1 0 10000|54
Chr1 10000 20000|99
Chr1 20000 30000|86
Chr1 30000 40000|60
Chr1 40000 50000|80
Chr1 50000 60000|112
Chr1 60000 70000|173
Chr1 70000 80000|173
Chr1 80000 90000|147
- To convert the counts to rates a simple shell command can be used. Just remember to also change the window size here if done previously.
# With std section and chr keyword (for use with Mutation-Simulator)
echo "std\nit None\nNone\n\nchr 1" > mutations.rmt && sed 's/|/\t/' mutations.windows.counts.bed | awk '{printf ("%s-%s sn %s\n", $2+1, $3, $4/10000) }' >> mutations.rmt
# Without (For further processing)
sed 's/|/\t/' mutations.windows.counts.bed | awk '{printf ("%s-%s sn %s\n", $2+1, $3, $4/10000) }' > mutations.rmtOften, data is available as multi sample VCFs. These can be used the same way as single-sample VCFs, but need some preprocessing.
- Extract only the desired sample.
bcftools view -s my_sample multi-sample.vcf.gz > only_my_sample.vcf- Remove remaining positions of other samples that are not covered in the extracted sample or equal to the reference sequence.
grep -vP '\t\./\.' only_my_sample.vcf | grep -vP '\t0/0' > only_my_sample_filtered.vcfVCF files can contain multiple types of alternate alleles, for example SNPs,
insertions and duplications. The type of an allele is stated in the alternative
allele field ##ALT=<ID=type,Description=description>. To create an RMT file
containing the rate distributions for multiple types of mutations, the VCF has
to be separated into one VCF for each mutation type. Rates must be calculated
individually and then be combined into one RMT file.
- Filter the file for each desired type, using, for example, GATKs
select variantstool with the--selectTypeoption. For possible types to filter and their abbreviations see the VCF spec. In this example, we filter for SNPs, insertions and duplications, creating three output VCFs.
java -jar GenomeAnalysisTK.jar -T SelectVariants -R reference.fasta -V input.vcf -o onlySNPs.vcf -selectType SNP
java -jar GenomeAnalysisTK.jar -T SelectVariants -R reference.fasta -V input.vcf -o onlyINSERTIONSs.vcf -selectType INS
java -jar GenomeAnalysisTK.jar -T SelectVariants -R reference.fasta -V input.vcf -o onlyDUPLICATIONSs.vcf -selectType DUP- For each VCF, create an RMT file following the Simple example for VCF to RMT conversion workflow.
- Cut out the positions and rates.
cut -d " " -f 4 onlySNPs.rmt > positions.txt
cut -d " " -f 2,3 onlySNPs.rmt > SNPRates.txt
cut -d " " -f 2,3 onlyInsertions.rmt > insertionRates.txt
cut -d " " -f 2,3 onlyDuplications.rmt > duplicationRates.txt- Paste positions and rates together
paste -d " " positions.txt SNPRates.txt insertionRates.txt duplicationRates.txt > multiType.rmtMinimum and maximum length options also need to be added in a similar fashion, depending on the included mutation types.
As a basic use case example we want to block all genes listed in a GFF3 file
from mutating and only establish SNPs between genes at a specified rate. For
this we provide an example
script
that can be used as a template to derive scripts for other use cases. The basic
idea is to filter all GTF/GFF lines by the type column (3rd column) and extract
the start and end position (4th and 5th column) of all genes. These intervals
will then be blocked in the output RMT
files.
The script can be run with:
python3 gff_genes_2_rmt.py [gff_file] [rate]Mutation-Simulator only supports haploid mutations, although multiple VCFs from different runs of Mutation-Simulator can be merged to form polyploid mutations.
These tests were performed in ARGS mode on a single sequence consisting of a random selection of bases with the following parameters:
-sn 0.01 -in 0.01 -de 0.01 -du 0.01 -iv 0.01 -tl 0.01| Sequence length [Mbp] | Runtime [s] | Memory peak [GB] |
|---|---|---|
| 1 | 6.49 | <0.01 |
| 10 | 51.41 | 0.20 |
| 100 | 517.20 | 1.48 |
| 1000 | 5235.40 | 13.92 |
- Testing Platform: Ubuntu 20.04.3 LTS
- Processor: AMD EPYC 7452
- Mutation-Simulator version: 3.0.1
- Measuring tool: GNU time
The memory consumption is affected by the number of introduced mutations as well as the length of the largest sequence mutated. Parameters chosen reflect a rather intense mutation of the genome. Thus, even genomes with large chromosomes up to 1 Gbp in length can be processed on a desktop computer with 16 GB of RAM.
Kühl, M. A., B. Stich, and D. C. Ries. "Mutation-Simulator: fine-grained simulation of random mutations in any genome." Bioinformatics 37.4 (2021): 568-569.