CompChem Tools
Whilst there are number of Open Source computational toolkits and command-line tools they often present a step learning curve for new users. In an effort to provide a simpler environment to access these tools this page will highlight a series of Jupyter notebooks that users can use to run key computational studies that might be undertaken in a drug discovery project.
To use these notebooks you will need to have Jupyter and a number of Python libraries installed. The easiest way to do this is to use Anaconda. Anaconda is a modern package manager and seems to be becoming the preferred source of scientific software.
Everyone is welcome to contribute Jupyter notebooks, I'd certainly recommend reading "Ten simple rules for writing and sharing computational analyses in Jupyter Notebook" DOI which gives some great tips.
Whether you use notebooks to track preliminary analyses, to present polished results to collaborators, as finely tuned pipelines for recurring analyses, or for all of the above, following this advice will help you write and share analyses that are easier to read, run, and explore.
To run these tools you will need a little familiarity with using the command-line interface. On a Mac this is the Terminal app found in the Utilities folder in the Applications folder. If you want a quick introduction to the UNIX command line try Learn UNIX in 10 minutes
Install Conda using the instructions here https://www.anaconda.com/distribution/
Then in a terminal window type
conda install jupyter
conda install -c rdkit rdkit
conda install numpy
conda install scipy
conda install scikit-learn
conda install pandas
conda install matplotlib
conda install seaborn
You should now have all the components installed to run Jupyter notebook. To run a notebook you need to use the Terminal to navigate to the folder containing the notebook, use the unix command cd (change directory) followed by the path to the folder.
cd /Users/username/Projects/OpenSourceAntibiotics/UsingSmina
If you then type ls (List) you should a listing of all files within the folder.
ls
373ligand_only.pdb protein_minus_ligand.pdb
ConformationGenerationDocking.ipynb protein_plus_373ligand.pdb
asinexSelection.sdf
To start the Jupyter kernel in the Terminal type
Jupyter notebook
You should see the jupyter server start up in the Terminal and your web browser open showing the list of files in the folder.
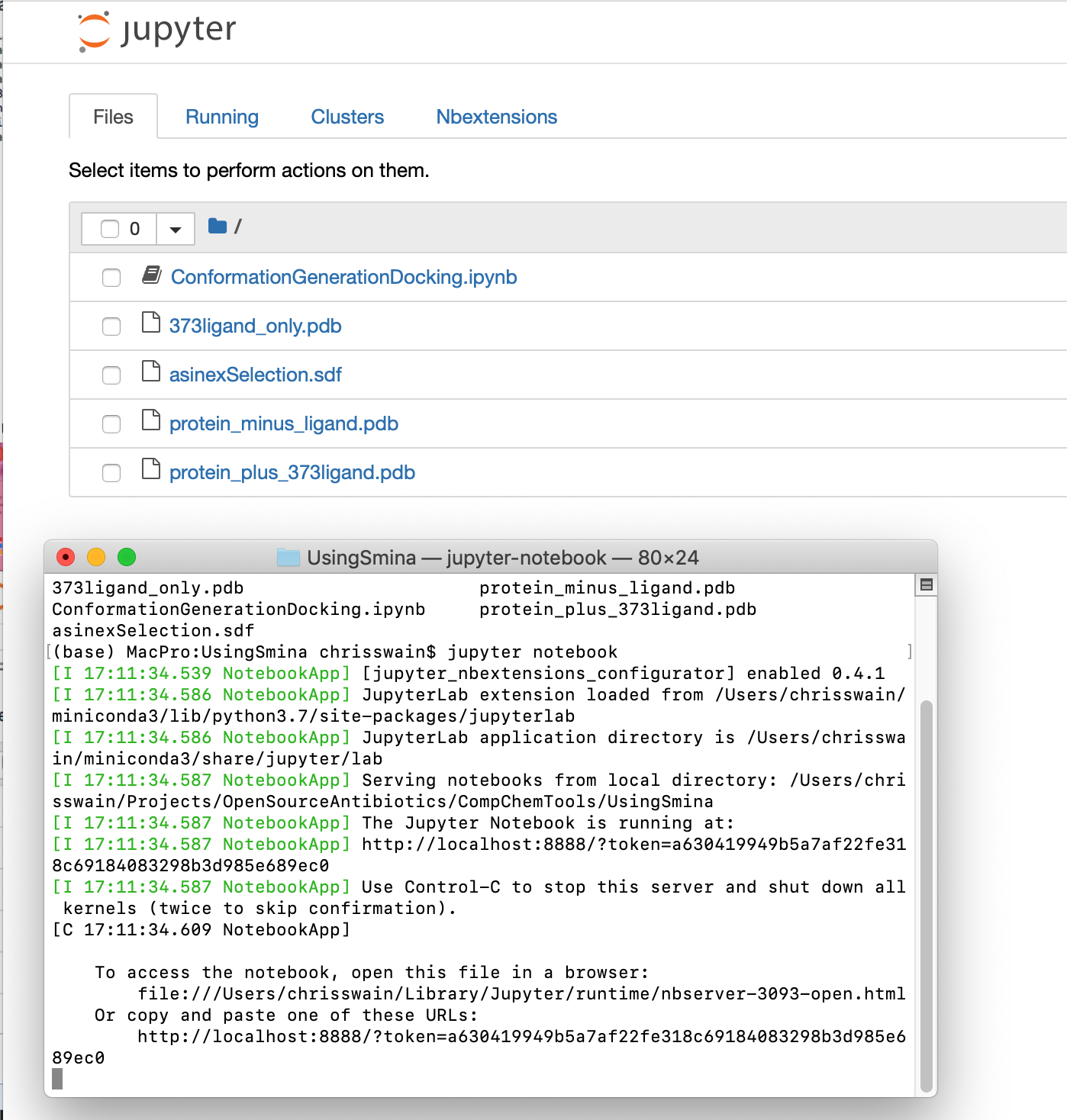
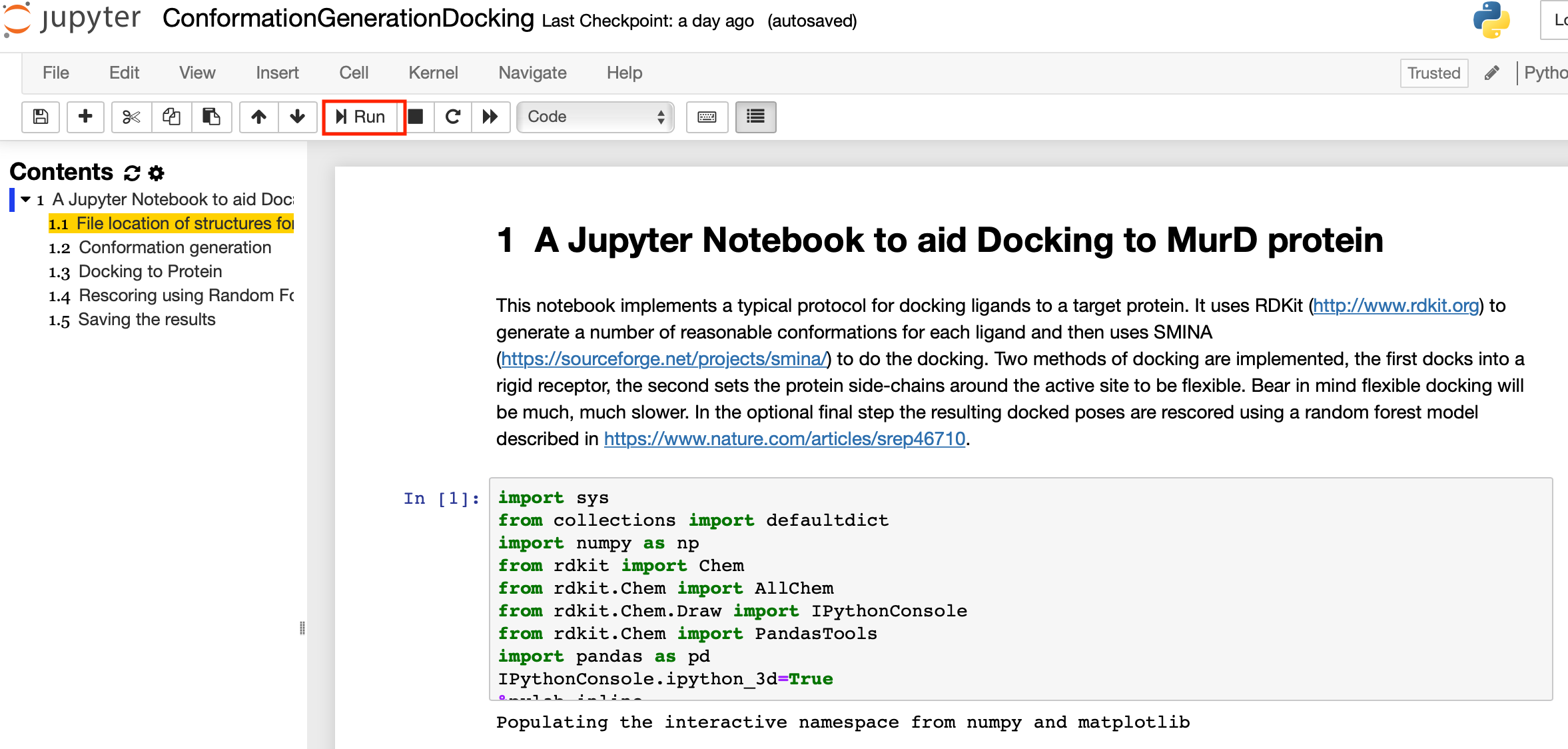
Now click on the link ConformationGenerationDocking.ipynb
This will open the notebook, you should now be able to step through the cells in the notebook by clicking the run arrow in the notebook menu bar.
SMINA (https://sourceforge.net/projects/smina/) is a command-line application for docking. Instructions for installation are on the website. By default it will be installed in
/usr/local/bin/smina.osx
After you have installed smina you need to give it permission to execute using the Terminal command
chmod +x /usr/local/bin/smina.osx
You can then check it is all working by typing in the Terminal
/usr/local/bin/smina.osx --help
and you should see the Smina help.
This notebook implements a typical protocol for docking ligands to a target protein. It uses RDKit (http://www.rdkit.org) to generate a number of reasonable conformations for each ligand and then uses SMINA (https://sourceforge.net/projects/smina/) to do the docking. Two methods of docking are implemented, the first docks into a rigid receptor, the second sets the protein side-chains around the active site to be flexible. Bear in mind flexible docking will be much, much slower. In the optional final step the resulting docked poses are rescored using a random forest model described in this publication DOI. You can read more details of the notebook here and you can download a folder containing the notebook and the necessary files here.
This notebook demonstrates how to get the structures and data from the master worksheet, then convert the SMILES to molecule objects using RDKit that allow some simple manipulations and visualisations. SMILES (Simplified Molecular Input Line Entry System) is a line notation (a typographical method using printable characters) for entering and representing molecules and reactions. https://www.daylight.com/dayhtml/doc/theory/theory.smiles.html You can read more details of the notebook here and you can download the notebook here.