A tool for finding tandem repeats in genomic assembles and reads.
Install the latest version of Miniconda (or Anaconda). If you already have either installed, skip to cloning the repository:
$wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
$chmod +x Miniconda3-latest-Linux-x86_64.sh
$./Miniconda3-latest-Linux-x86_64.sh
Restart your terminal to auto-enter your base conda environment.
Install git if it's not installed:
$sudo apt-get install git
Clone this repository into a directory of your choice:
$git clone https://github.com/rnhall/TANDEM.git
$cd TANDEM
In the main directory, you'll see environment.yml, this tells conda which dependencies to install.
We'll make a new conda environment and set it up with environment.yml.
$conda config --add channels conda-forge
$conda config --add channels bioconda
$conda create --name tandem
$conda activate tandem
$conda env update -f environment.yml
Once the environment is done being set up, try running TANDEM on a test file:
(You may have to enable the file to be run)
$chmod +x tandem.py
$./tandem.py -genome test_sequence.fasta
You may also need to install libgtk-3-0:
$sudo apt install libgtk-3-0
TANDEM, in its present form can be split into two major steps: first, a coarse search for low-complexity regions (complexity_masker) of the genome provides a subset of sequences which is then further analyzed by a graph-based repeat finder (cycle_finder).
 First, complexity_masker slides a window across each chromosome or contig in your genome file. It then calculates a k-mer complexity score for each bin. After calculating the k-mer complexity score across all provided sequences, it identifies regions of the genome where the k-mer complexity fell below the designated cutoff (default is 0.8).
First, complexity_masker slides a window across each chromosome or contig in your genome file. It then calculates a k-mer complexity score for each bin. After calculating the k-mer complexity score across all provided sequences, it identifies regions of the genome where the k-mer complexity fell below the designated cutoff (default is 0.8).
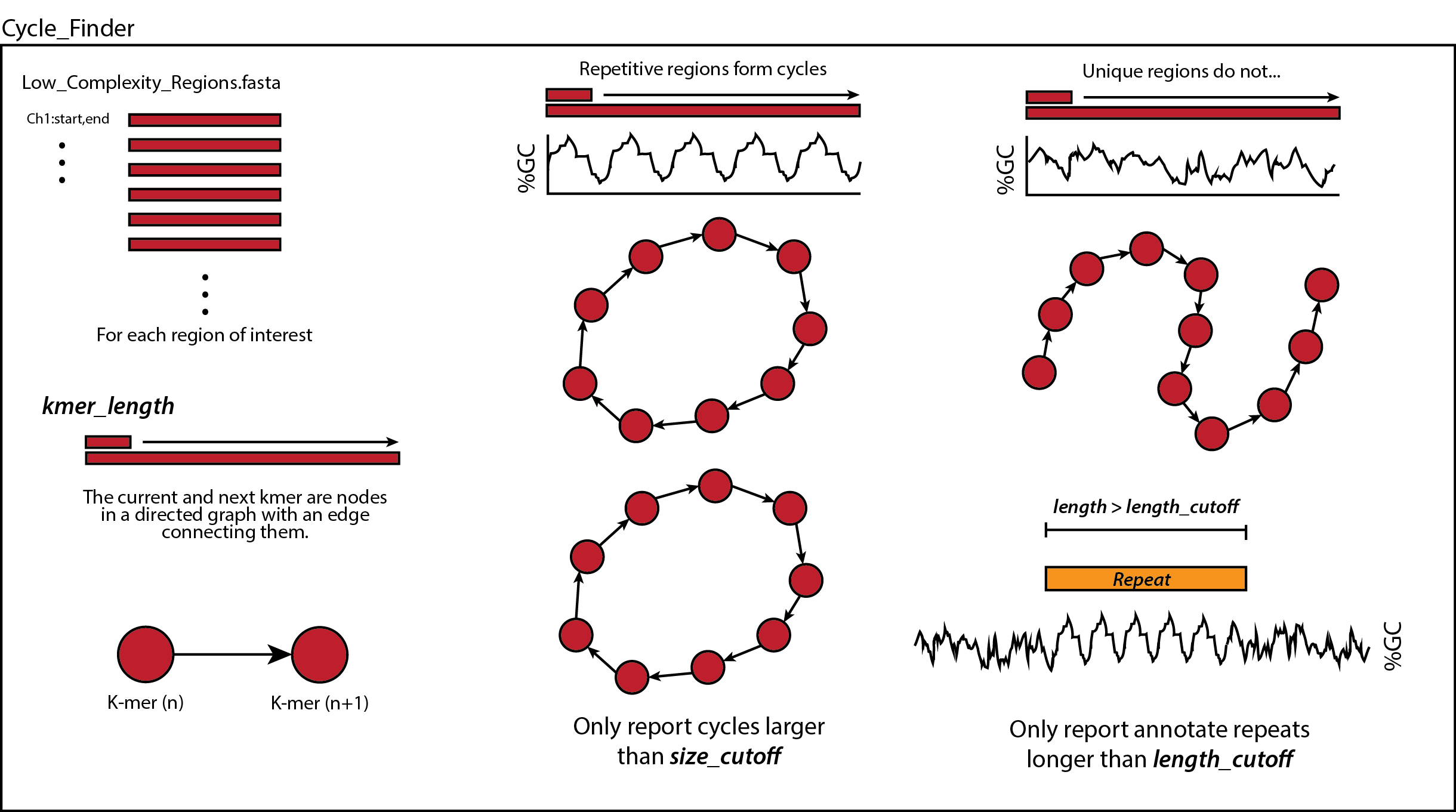 Then, each low-complexity region is further analyzed by cycle_finder. This too converts each region of interest into a directed graph of k-mers. Each node represents a k-mer, and each edge represents a k-mer transitioning from one to another. The edges are weighed by how often that transition is observed, normalized by the sum of all outgoing edge weights, then the -log is taken to ensure that frequent transitions are small positive values and infrequent transitions are large positive values. This step is important since the consensus sequence is determined to be the cycle which maximizes the number of nodes visited while minimizing the sum of edge weights. Importantly, repetitive sequences produce cyclic graphs while unique regions do not. Once all regions have been analyzed and repetitive sequences have been found, only repeats that are larger than size_cutoff and occupy space on the genome larger than length_cutoff are reported.
Then, each low-complexity region is further analyzed by cycle_finder. This too converts each region of interest into a directed graph of k-mers. Each node represents a k-mer, and each edge represents a k-mer transitioning from one to another. The edges are weighed by how often that transition is observed, normalized by the sum of all outgoing edge weights, then the -log is taken to ensure that frequent transitions are small positive values and infrequent transitions are large positive values. This step is important since the consensus sequence is determined to be the cycle which maximizes the number of nodes visited while minimizing the sum of edge weights. Importantly, repetitive sequences produce cyclic graphs while unique regions do not. Once all regions have been analyzed and repetitive sequences have been found, only repeats that are larger than size_cutoff and occupy space on the genome larger than length_cutoff are reported.
The program provides outputs in the form of a .fasta and .gtf file containing the identified low-complexity regions, a .fasta and .gtf of assembled repeat monomers and their locations in the genome, and finally a .fasta containing a list of all unique consensus repeat monomers.
For details on required and optional parameters, use the help functionality:
$./tandem.py -h
The minimal use case will use the default parameters:
$./tandem.py -genome {path/to/genome.fasta}