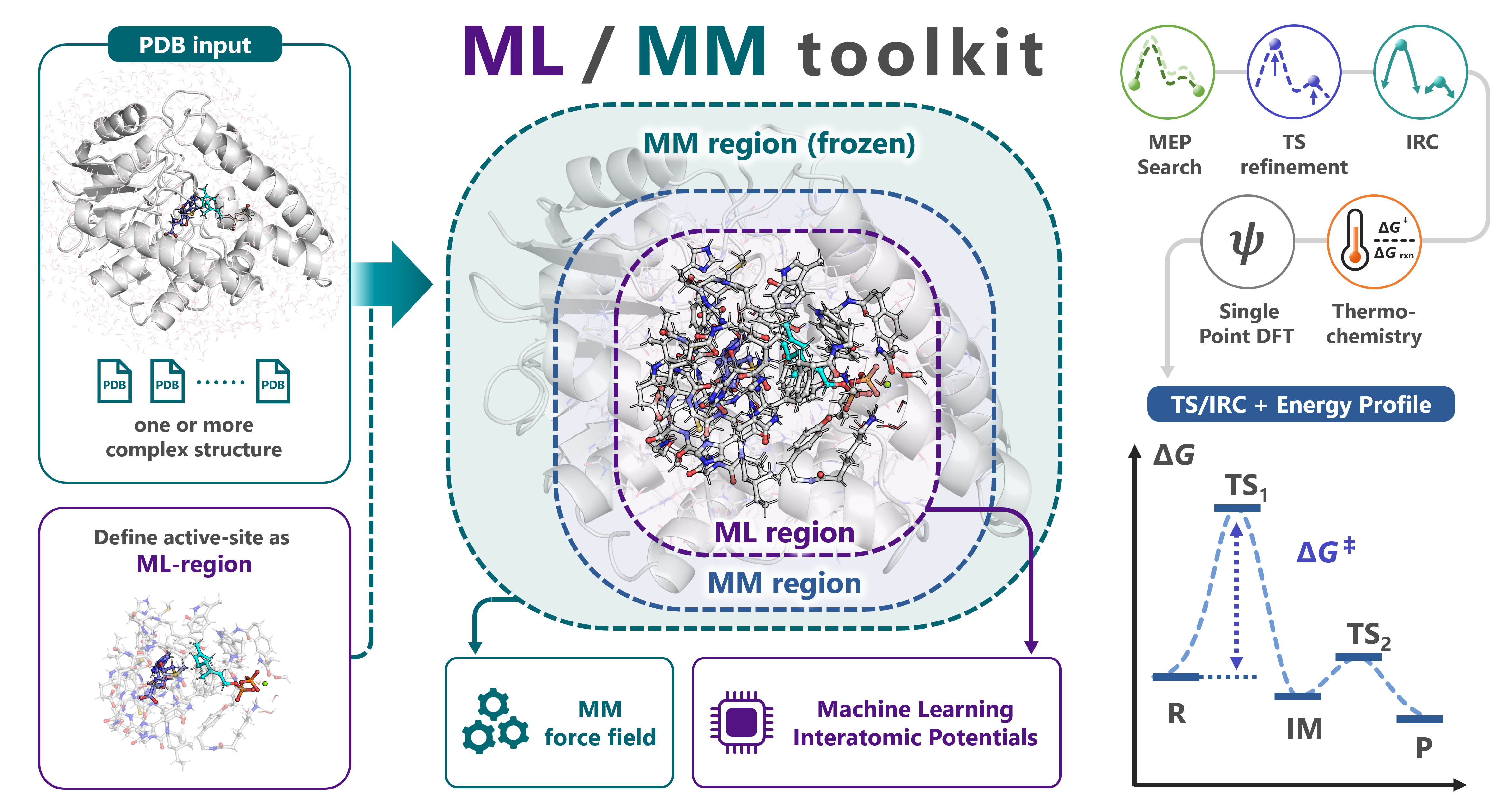
mlmm-toolkit is an open-source CLI for ML/MM ONIOM analyses of enzymatic reactions. It replaces the QM region of conventional QM/MM with a machine-learning interatomic potential (MLIP, default: UMA) while keeping the surrounding protein under an analytical Amber force field (hessian_ff), and chains MM parametrization → ML-region selection → MEP search → TS optimization → IRC → thermochemical correction → DFT single-point in one command. A link-atom boundary handles amino-acid residues straddling the ML/MM cut, and a microiteration scheme makes TS optimization and Hessian-based methods tractable on ~10 000-atom systems.
Test a reaction mechanism in a single command:
# Multi-structure MEP (R + P endpoints → MEP, with TS optimization + thermo)
mlmm all -i R.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' --tsopt --thermoFor scan-mode on a single structure and the bundled methyltransferase walk-through, see examples/. Each stage is also exposed as an individual subcommand.
Prerequisites: input PDBs must already contain hydrogens; multiple PDBs must share the same atoms in the same order (only coordinates differ).
mlmm allrunsmm-parmautomatically. Match-l RES:CHARGEto the H count actually present (e.g. SAM with 23 H =SAM:1cation, 22 H =SAM:0neutral) — full input-prep checklist in docs/getting-started.md.
| Tool | Use case |
|---|---|
| pdb2reaction | Pure-MLIP reaction paths for cluster models and small molecules from PDB / XYZ / GJF. |
| uma_pysis | Lightweight YAML-driven UMA–pysisyphus interface for quick/exploratory reaction-mechanism studies (GS / TS / IRC / ΔG). |
mlmm-toolkitbundles a GPU-optimized pysisyphus fork that is not compatible with upstream pysisyphus — do not install it into an environment that already has upstream pysisyphus.
- Getting Started · Concepts · Installation · Troubleshooting
- Python API · CLI Conventions · YAML Reference · JSON Output Schema
- Full command index: docs/index.md
| Component | Requirement |
|---|---|
| OS / Python | Linux recommended; native Windows unsupported (AmberTools/tleap unavailable). Python >= 3.11. |
| GPU / CUDA / VRAM | NVIDIA GPU, CUDA >= 12.6 (12.8+ recommended; required for RTX 50-series). 8 GB+ VRAM recommended. |
| RAM / Disk | 32 GB+ RAM recommended; 20 GB free disk for the conda env, AmberTools, UMA cache, and artifacts. |
Required external tools: AmberTools (tleap) and pdbfixer — conda install -c conda-forge ambertools pdbfixer -y. CPU-only execution works for setup commands but is 10–100× slower for any ML/MM dynamics or Hessian step. Full requirement and tuning details: docs/getting-started.md#installation.
# 1. New env + AmberTools + CUDA-enabled PyTorch
conda create -n mlmm-toolkit python=3.11 -y && conda activate mlmm-toolkit
conda install -c conda-forge ambertools pdbfixer -y
pip install torch --index-url https://download.pytorch.org/whl/cu129
# 2. mlmm-toolkit (editable from a local clone, or `pip install mlmm-toolkit`)
pip install -e .
# 3. Authenticate Hugging Face once (only required for the default UMA backend)
# Accept the FAIR Chemistry License v1 at https://huggingface.co/facebook/UMA, then:
hf auth login # interactive
# OR: export HF_TOKEN=hf_xxx && hf auth login --token "$HF_TOKEN" # CI / HPCAvoid AmberTools conflicts: on clusters with a system AmberTools module loaded, run
module unload amberbefore installing to prevent a ParmEd conflict with the conda-installed AmberTools.
Optional extras (install only what you need):
| Extra | Adds |
|---|---|
[orb] / [aimnet] |
Orb / AIMNet2 MLIP backend — not HF-gated |
[dft] |
PySCF + GPU4PySCF single-point DFT (--dft / mlmm dft) — practical to ~500 ML-region atoms |
[mcp] |
Model Context Protocol server (mlmm-mcp) for agent clients |
[pdbfixer] |
PDBFixer extra (alternative to the conda install above) |
The MACE backend (-b mace) is not a pip extra: mace-torch pins e3nn==0.4.4, which conflicts with fairchem-core's e3nn>=0.5 (UMA), so it needs a dedicated environment — pip uninstall -y fairchem-core && pip install mace-torch (see docs/getting-started.md#installation).
CUDA module-load recipes, alternative-backend installs, DMF / cyipopt, Plotly Chromium, and HPC job-script templates: docs/getting-started.md and docs/device-hpc.md.
For most systems the only hard requirement is a PDB with explicit hydrogens (at the intended protonation state). mlmm all then builds the MM topology, selects the ML region, and runs the whole pipeline in one command — see Quick Examples for the three input modes (multi-structure R → P, single-structure scan, TS-only). The preparation steps below are optional.
-
Build a structural model of the complex. Download coordinates from the Protein Data Bank. If an experimental structure is not available, use structure-prediction programs such as AlphaFold3, Boltz2, or Chai; docking programs; or GUI software such as PyMOL. Add hydrogens at the intended protonation state (or let
mm-parm --add-h --ph 7add them). For multi-structure (R → P) runs, every PDB must share the same atoms in the same order. -
(Optional) Build the MM topology yourself — it is automatic by default.
mlmm all(viamlmm mm-parm) generates the Amber.parm7/.rst7from the PDB automatically; unknown residues (ligands, cofactors) are parameterized with GAFF2 / AM1-BCC — pass formal charges with-l 'RES:CHARGE'. Build the topology by hand when it helps — a custom force field, special solvation, or a system the automatic route cannot handle — then pass it with--parm. To mimic aqueous conditions, solvate the complex and remove water molecules beyond ~6 Å (see the OpenMM cookbook / tleap).Note: elemental information (columns 77–78) is omitted in PDB files generated by tleap. Use
mlmm add-elem-infoto fix this. -
(Optional) Define the ML region yourself.
mlmm allextracts the ML region from-c/--centerand-r/--radiusautomatically. To define it yourself instead, build an ML-region PDB — withmlmm extractor any molecular viewer — and feed it tomlmm all(or the per-stage subcommands) with--model-pdb; this skips the automatic extraction:mlmm extract -i complex.pdb -c 'SAM,GPP' -r 6.0 -l 'SAM:1,GPP:-3' -o ml_region.pdb
Important: atom order, residue names, and residue numbers must match between the full PDB and the ML-region PDB. (In PyMOL, tick "Original atom order" when exporting.)
# Multi-structure MEP (R + P → MEP, with TS + thermo + DFT)
mlmm all -i R.pdb P.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' --tsopt --thermo --dft
# Scan mode (single structure → staged bond scans → MEP)
mlmm all -i R.pdb -c 'SAM,GPP' -l 'SAM:1,GPP:-3' \
--scan-lists "[('SAM 359 CS1','GPP 360 C8',1.3)]"
# TS-only validation (existing TS candidate)
mlmm tsopt -i TS_candidate_layered.pdb --parm complex.parm7 -q 1 --opt-mode gradFor Gaussian-ONIOM / ORCA-QM/MM round-trips use oniom-export / oniom-import. Per-stage walkthrough (mm-parm → extract → define-layer → opt → path-search → tsopt → freq → irc → dft): docs/getting-started.md and docs/quickstart-all.md. Working scripts (methyltransferase + toy_system): examples/.
A run writes its deliverables to --out-dir (default ./result_all/):
segments/seg_NN/{reactant,ts,product}.pdb— the canonical R / TS / P structures to citemep.pdb/mep_trj.xyz— the merged reaction path;energy_diagram_MEP.png— barrier diagramsummary.log(human-readable) /summary.json(machine-readable)- Reusable inputs for follow-up runs:
ml_region.pdb(--model-pdb),mm_parm/*.parm7(--parm),layered/(B-factor-annotated full-system PDBs)
Pipeline scratch lives under _work/ (safe to delete). Full layout and filename conventions: docs/output-layout.md.
| Subcommand | Role | Doc |
|---|---|---|
all (default) |
End-to-end: mm-parm → extract → MEP → TS → IRC → freq → DFT |
all |
mm-parm |
Generate parm7/rst7 via AmberTools | mm-parm |
extract |
Extract active-site pocket | extract |
define-layer |
Assign 3-layer ML/MM B-factor encoding | define-layer |
add-elem-info / fix-altloc |
Repair PDB element columns / resolve altlocs | add-elem-info · fix-altloc |
opt / tsopt |
Geometry / TS optimization | opt · tsopt |
path-opt / path-search |
MEP via GSM/DMF; recursive refinement | path-opt · path-search |
scan / scan2d / scan3d |
1D / 2D / 3D bond-distance scans | scan · scan2d · scan3d |
freq / irc |
Vibrational analysis + thermo / IRC (EulerPC) | freq · irc |
dft / sp |
Single-point DFT / single-point ML/MM ONIOM | dft · sp |
bond-summary |
Compare structures, report bond changes | bond-summary |
trj2fig / energy-diagram |
Energy plot / R→TS→P diagram | trj2fig · energy-diagram |
oniom-export / oniom-import |
Gaussian ONIOM / ORCA QM/MM round-trip | oniom-export · oniom-import |
3-layer system (ML / Movable-MM / Frozen-MM, B-factor encoded), link-atom treatment, units (eV·Å in core / Ha·Bohr in pysisyphus CLI): docs/concepts.md. Python API (MLMMCore, MLMMASECalculator, pysisyphus mlmm calculator): docs/python-api.md.
mlmm --help # top-level
mlmm <subcmd> --help # core options
mlmm <subcmd> --help-advanced # full option setIssues: https://github.com/t-0hmura/mlmm_toolkit/issues.
@article{ohmura2025mlmm,
author = {Ohmura, Takuto and Inoue, Sei and Terada, Tohru},
title = {ML/MM Toolkit -- Towards Accelerated Mechanistic Investigation of Enzymatic Reactions},
year = {2025}, journal = {ChemRxiv}, doi = {10.26434/chemrxiv-2025-jft1k}
}Agent Skills for Claude Code / Codex / Cursor etc. in skills/ — copy into your project's skill location (e.g. .claude/skills/) to let an agent drive mlmm-toolkit workflows and subcommands.
- MACE + UMA cannot coexist (
e3nnversion conflict). Use separate conda envs. - DFT single-point is practical to ~500 ML-region atoms; larger regions incur high computational cost.
- ORB backend sometimes converges TS with extra soft imaginary modes — prefer UMA / MACE for clean single-saddle spectra.
- CPU-only execution is 10–100× slower than GPU; AmberTools (
tleap) is required formm-parm.
Issues and pull requests are welcome — see CONTRIBUTING.md.
GNU General Public License v3 (GPL-3.0).