

This is the homepage of TransPhylo, a software package that can reconstruct infectious disease transmission using genomic data. The input is a dated phylogeny, where leaves correspond to pathogens sampled from the known infected hosts. The main output is a transmission tree which indicates who infected whom, including the potential existence of unsampled individuals who may have acted as missing transmission links. TransPhylo works by colouring the branches of the phylogeny using a separate colour for each host, sampled or not. Each section of the tree coloured in a unique colour represents the pathogen evolution happening within a distinct host. Changes of colours on branches therefore correspond to transmission events from one host to another.
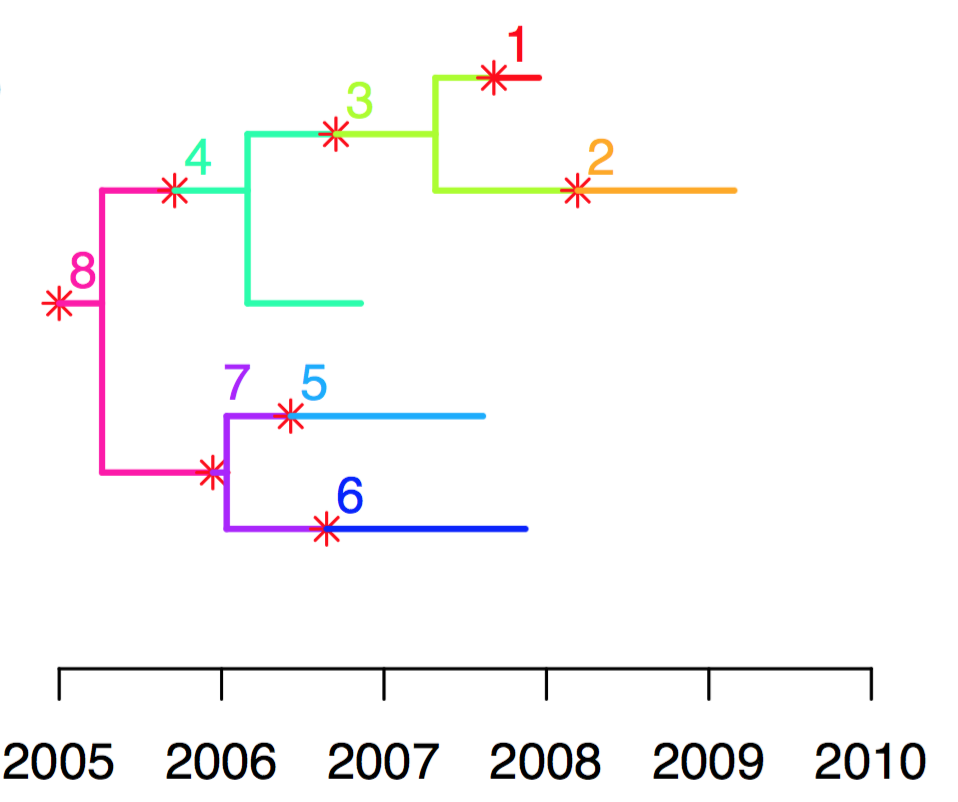
For example, in the tree below, the outbreak started with the unsampled host 8, who transmitted to sampled host 4, who transmitted to unsampled host 3, who transmitted to both sampled hosts 1 and 2. Host 8 also transmitted to unsampled host 7 who transmitted to both sampled hosts 5 and 6 .
For a more formal description of TransPhylo, see the following paper:
Didelot, Fraser, Gardy and Colijn (2017) Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks Molecular Biology and Evolution, 34:997-1007 https://academic.oup.com/mbe/article/34/4/997/2919386
You can install TransPhylo in R using the following command:
devtools::install_github('xavierdidelot/TransPhylo')
See the TransPhylo tutorial at https://xavierdidelot.github.io/TransPhylo/articles/TransPhylo.html. This tutorial is also available within the R package as a vignette. The TransPhylo reference manual is available at https://xavierdidelot.github.io/TransPhylo/reference/index.html.
If you need assistance using TransPhylo, you can get in touch by
emailing xavier.didelot@gmail.com
If you are looking for the older Matlab version of TransPhylo described in Didelot et al (2014) Molecular Biology and Evolution 31:1869-1879, please note that this has now been moved to the repository TransPhyloMatlab, available at https://github.com/xavierdidelot/TransPhyloMatlab