This is a workflow pipeline for analyzing Synechococcus genomes and creating a Phylogenetic Tree using the guide given by astrobiomike
- GToTree
If you would like to use the envionment used to run this analysis I have already supplied it on the .yml file. All you have to do is to run the code below within this repository:
conda env create -f gtotree_env.yml
- Genomes to be worked on
- Target genes to be used for the pylogenetic tree (Reference)
For this example we used Synechococcus that was already given by the site.
curl -L -o syn-gtotree-example.tar.gz https://ndownloader.figshare.com/files/23629763
Were the commands did the following actions:
- curl ; Transfer data from a server
- L ; Define the site from were we'll obtain the data
- o ; Define where we want to store the file or to name is at we require
So based on the command we are downloading the dataset and renaming it syn-gtotree-example.tar.gz to be a compressed archive
To extract the files from the compressed archive we'll use the tar command
tar -xvzf syn-gototree-example.tar.gz
Were the commands did the following actions:
- x ; extract the files from the archive
- v ; show the progress of the extraction
- z ; filter the files through gzip
- f ; create files with given filenames
This will create a folder with the following files
- Sequence Genomes Files (fasta files)
- NCBI Assembly accessions (ref-syn-accs.txt)
To input the data we need to list them in a txt file
ls *.fa > our-genome-fasta-files.txt
Were the commands did the following actions:
- ls ; create a list
- *.fa ; this will include every file that has a name terminating with fasta type file
GToTree includes several HMM target genes sets which is beneficial for this example as Synechococcus is a Cyanobacteria which is included on GToTree
##Running GToTree To run GToTree we'll use the code as presented below which will download the amino-acid sequences (ref-syn-accs.txt), identify the target genes on our fasta files (our-genome-fasta-files.txt), filter them and complete genes where necessary, align them to each individual gene-set, trims them and cocatenate them to obtain the necessary data to develop our philogenetyc tree.
GToTree -a ref-syn-accs.txt -f our-genome-fasta-files.txt -H Cyanobacteria \
-t -L Species -j 4 -o Syn-GToTree-out
Where the commands did the following actions:
- GToTree ; Start running the phylogenomic workflow to do the analysis
- a ; Indicates which file contains the file assembly accessions (ref-syn-accs.txt ; Obtained from downloaded compressed file)
- f ; Indicates the genomes we obtained as fasta files (our-genome-fasta-files.txt ; Obtained listing the fasta files obtained from compressed file)
- H ; The target genes that will be aligned to our selected genome (Cyanobacteria ; Obtained from the integrated HHM data on GToTree)
- \ ; To continue the code in a different line for better visibility
- t ; To use TaxonKit to add the lineage information for the tree
- L ; Allows to set the tree to different type of data, in this case it will be set to "Species" (This will include genus, species and strain designations in NCBI data)
- j ; Sets the quantity of alignments that the workflow will perform in parallel (in this case it will run four at the same time)
- o ; Set the name of the directory (In this case it will be Syn-GToTree-out)
To make our visual more appealing, we'll do a color-mapping file to highlight our genomes. The genomes that we'll need to higlight will be our sequence genomes so we'll use our-genome-fasta-files.txt to obtain a list of the names.
cut -f 1 -d "." our-genome-fasta-files.txt > labels-to-color.txt
Where the commands did the following actions:
- cut ; Remove
- f ; Remove by fields (1; It will cut at the first delimiter)
- d ; Determines the delimiter (every period will be a delimiter)
- All the functions will be applied to the our-genome-fasta-files.txt and the resulting data will be stored in labels-to-color.txt
This file needs to be compatible with iTol and thus we'll run the following code
gtt-gen-itol-map -g labels-to-color.txt -o iToL-colors.txt
The following files need to be downloaded to the computer you're using to upload then to : https://itol.embl.de/
- Syn-GtoTree-out.tre
- iToL-colors.txt
After clicking the link showed above we'll see the following window
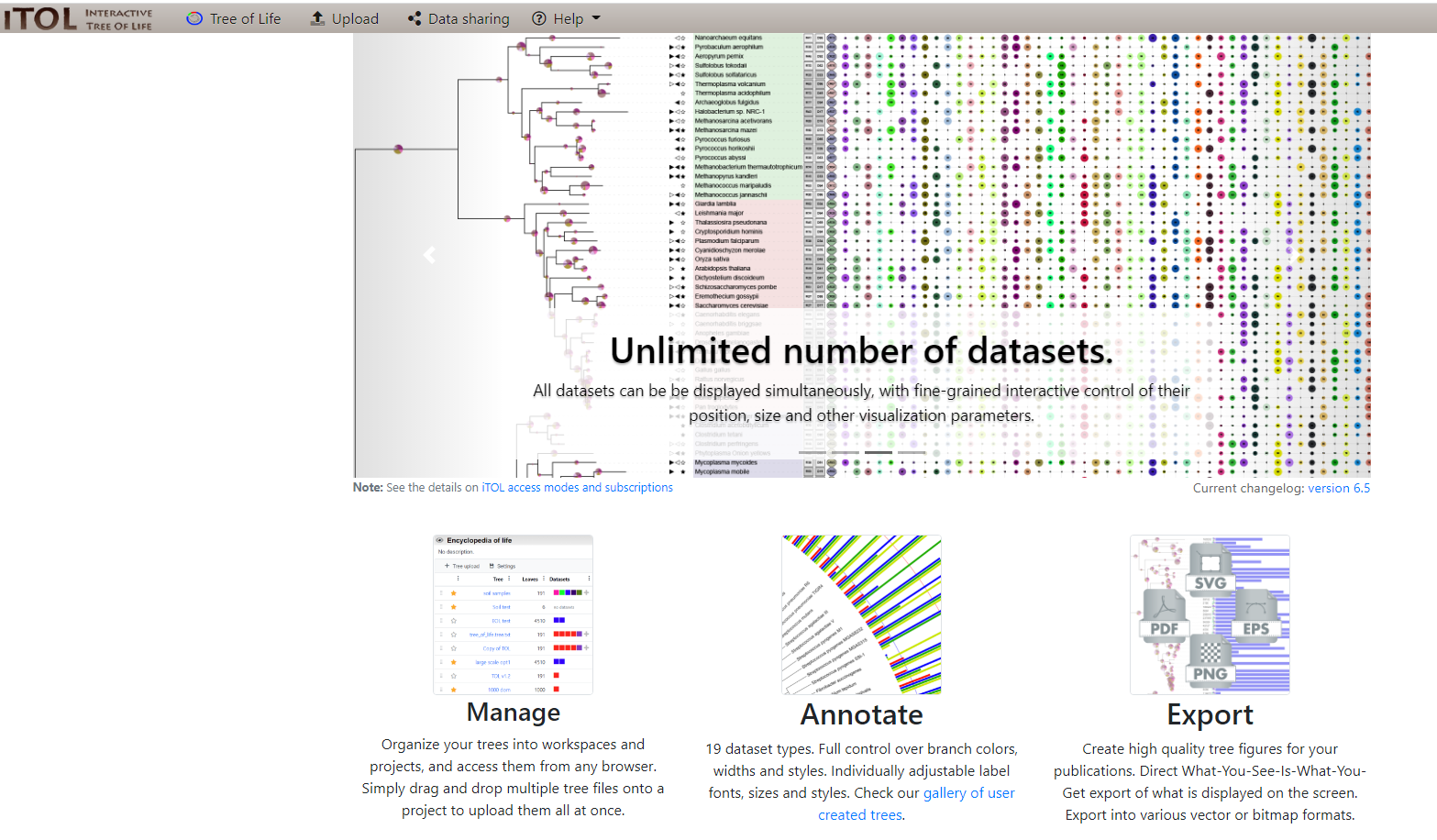
Where we'll select the Upload option on the menu above and input our Tree name (Synechococcus) and input Syn-GtoTree-out.tre
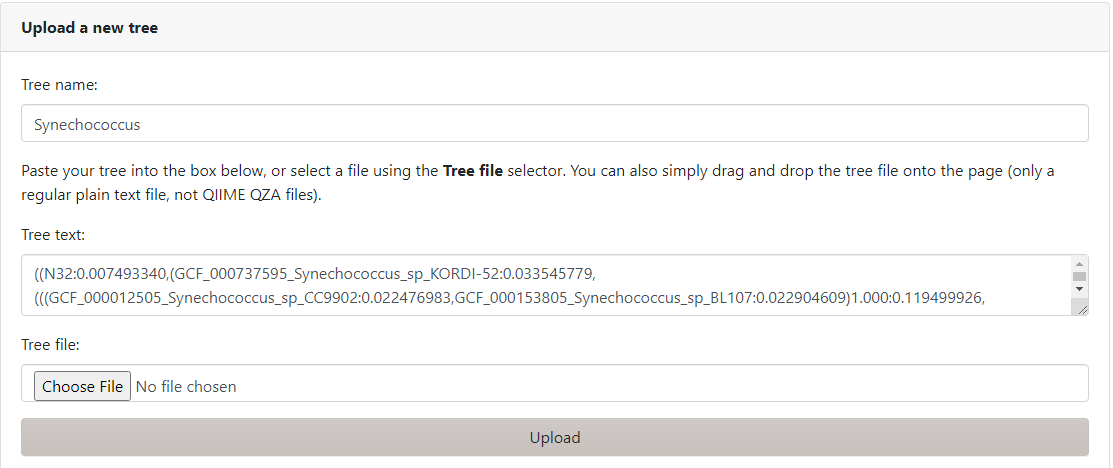
It will give us a draft which we can modify, for our purpose we'll Reroot the tree structure to the genome Gloeobacter violaceus
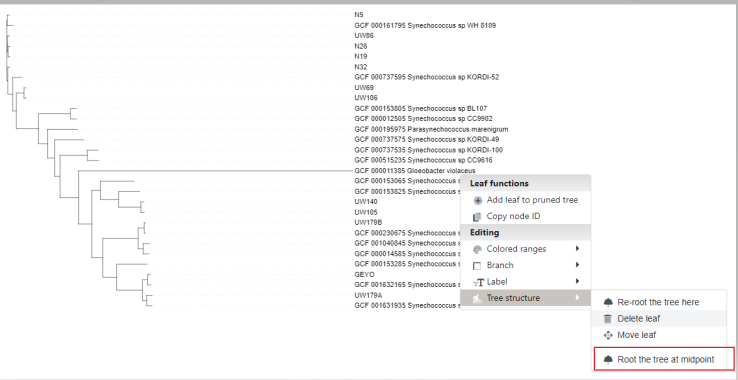
And finally we'll drop our iTOL color file to highlight our genomes
As a final result below i showed the result of following Phylogenetic Tree Guide.
