
iGDP-rc can work as a "positive or negative filter" to obtain target rumen ciliate sequences from genomic sequencing data containing various contaminants by integrating homology search, telomere reads-assisted and clustering approaches.

- Issues, bug reports and feature requests: GitHub issues
- Contact: Fei Xie (xiefei_njau@163.com); Chuanqi Jiang (jiangchuanqi@ihb.ac.cn)
- Citation: 1. Chuanqi Jiang, Guangying Wang, Jing Zhang, Siyu Gu, Xueyan Wang, Weiwei Qin, Kai Chen, Dongxia Yuan, Xiaocui Chai, Mingkun Yang, Fang Zhou, Jie Xiong, Wei Miao (2023). iGDP: An integrated Genome Decontamination Pipeline for wild ciliated microeukaryotes. Molecular Ecology Resources. 23, 1182–1193 (2023).
# mmseqs2 (>=v13.45111)
$ conda install -c bioconda mmseqs2
# bwa (>=v0.7.17)
$ conda install -c bioconda bwa
# samtools (>=v1.7)
$ conda install -c bioconda samtools
# metabat2 (>=v2.12.1)
$ conda install -c bioconda metabat2
$ git clone https://github.com/GWang2022/iGDP.git
# give executable permission to all scripts in iGDP scripts directory
$ chmod a+x iGDP/scripts/*pl
# add iGDP-rc scripts directory to your PATH environment variable
$ echo 'PATH=$(pwd)/iGDP/scripts/:$PATH' >> ~/.bashrc
$ source ~/.bashrc
# Usage: mmseqs databases <name> <o:sequenceDB> <tmpDir> [options]
# Downloading NR database named with prefix 'NRdb' in your working directory using the following command
$ mmseqs databases NR NRdb tmpDir
Tip: You can creat your own database for homology search using mmseqs createdb module. For more details, see mmseqs.
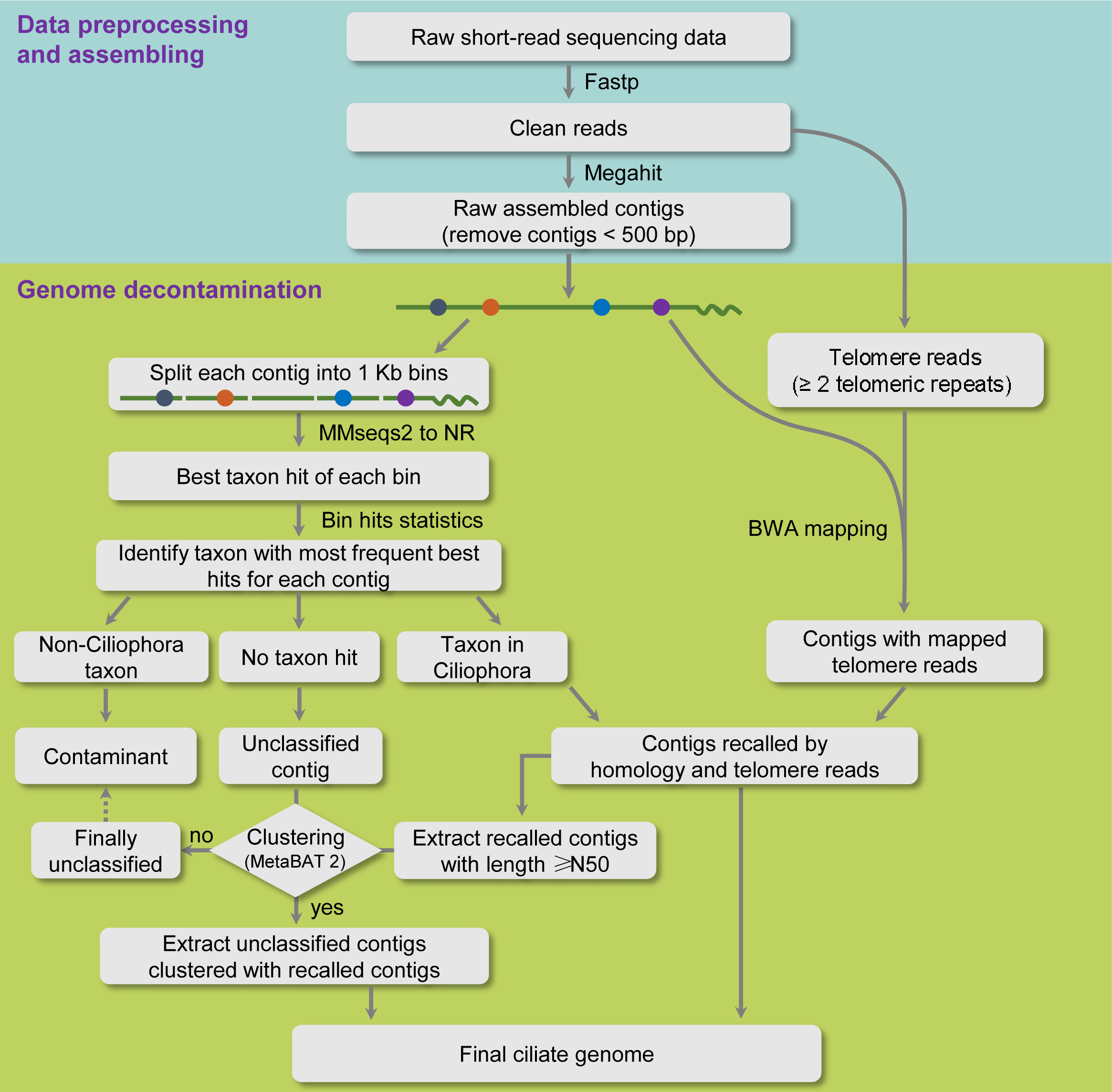
$ iGDP_homology_search.pl -i <input.contigs.fa> -o <output_dir> -d <mmseqs_DB> [options]
options:
-i <required>: input assembled contigs [.gz or uncompressed]
-o <required>: output directory [e.g. homology_search]
-d <required>: database for mmseqs search
-rank [optional]: target taxonomic space of homology search [format, rank:taxon; rank must be phylum/class/order/family/genus/species and taxon begins
with a capital letter; default: phylum:Ciliophora]
-b [optional]: bin size [contig is cut to -b bp for homology search; default: 1000]
-s [optional]: mmseqs seach sensitivity [1.0 faster; 4.0 fast; 7.5 sensitive; default: 5.7]
-t [optional]: number of threads used for mmseqs [default: 72]
-T [optional]: translation table of the target genome [default: 6 for ciliates]
$ iGDP_telomere_reads.pl -i <input.contigs.fa> -o <output_dir> -r1 <reads1> -r2 <reads2> [options]
options:
-i <required>: input assembled contigs [.gz or uncompressed]
-o <required>: output directory [e.g. telomere_reads]
-r1 <required>: read1 input file name [.gz or uncompress]
-r2 <required>: read2 input file name [.gz or uncompress]
-u [optional]: 5' to 3' telomeric repeat unit of the target genome [default: CCCCAA for Tetrahymena species]
-b [optional]: threads for bwa mem [default: 8]
-s [optional]: threads for samtools view [default: 8]
$ iGDP_clustering.pl -i <input.contigs.fa> -o <output_dir> -r1 <reads1> -r2 <reads2> [options]
options:
-i <required>: input assembled contigs [.gz or uncompressed]
-o <required>: output directory [e.g. clustering]
-r1 <required>: read1 input file name [.gz or uncompress]
-r2 <required>: read2 input file name [.gz or uncompress]
-b [optional]: threads for bwa mem [default: 8]
-s [optional]: threads for samtools view [default: 8]
Tip: Running iGDP_clustering.pl must be after implementing iGDP_homology_search.pl and iGDP_telomere_reads.pl programs.
This mode directly selects ciliate sequences as the target genome.
Please enter the iGDP/ directory after downloading iGDP and NR protein database.
You will see three files in the example/ directory:
- The file
assemly.fa.gzis a contaminated genome assembly. - The files
read1.fq.gzandread2.fq.gzare paired-end short-read sequencing data for the above genome.
Enter the example/ directory and implement the following command lines:
$ iGDP_homology_search.pl -i assemly.fa.gz -o homology_search -d {path_to_NR}/NRdb
$ iGDP_telomere_reads.pl -i assemly.fa.gz -o telomere_reads -r1 read1.fq.gz -r2 read2.fq.gz
$ iGDP_clustering.pl -i assemly.fa.gz -o clustering -r1 read1.fq.gz -r2 read2.fq.gz
Then the follwong data files will be created and deposited in the example/ directory:
-
The files
homology_search.homology.recall.contigs,telomere_reads.telo_reads.recall.contigsandclustering.contigscontain contig IDs obtained byiGDP_homology_search.pl,iGDP_telomere_reads.plandiGDP_clustering.plprograms, respectively; -
The folders
homology_search/,telomere_reads/andclustering/contain intermediate data files generate by the above commands. -
The file
final_genome.fais the final genome after contamination removal.
This mode first selects sequences from all non-Ciliophora contaminants and then keep the rest as the target genome. Compared with positive filtering, the obtained genome by this mode usually has higher completeness but lower precision.
After run iGDP_homology_search.pl and iGDP_telomere_reads.pl as above, implement the following command line:
$ iGDP_clustering_negative.pl -i assemly.fa.gz -o clustering_negative -r1 read1.fq.gz -r2 read2.fq.gz
- The file
final_genome.negative.fais the final genome after contamination removal.
- 2022/10/14
- intergate clustering program into iGDP
- add
-rankoption allowing user to set the homology search space for the target species.
- 2023/01/25
- add
negative filtering modeinto iGDP. This mode is suitable to genomic data without contamination from other ciliates such as single-cell sequencing data.
- add