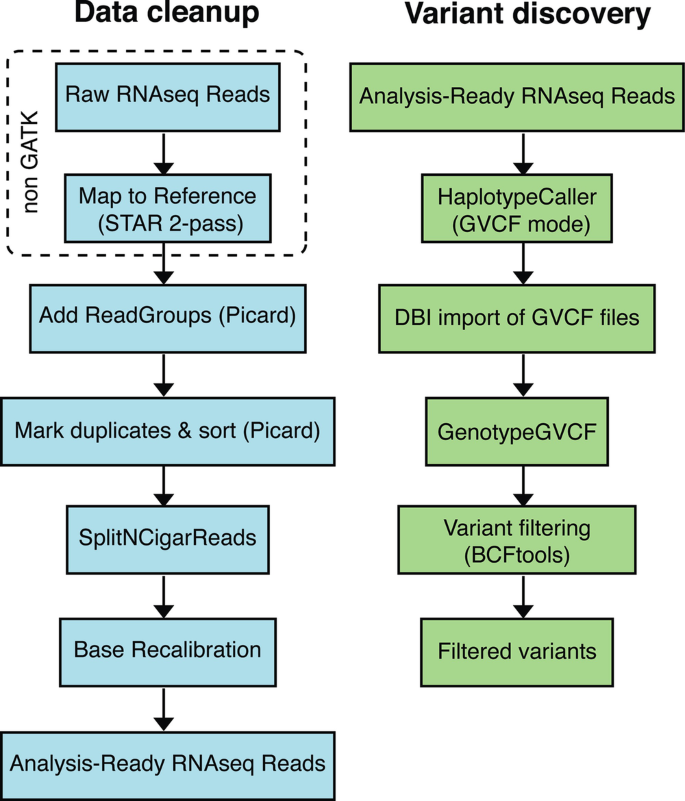
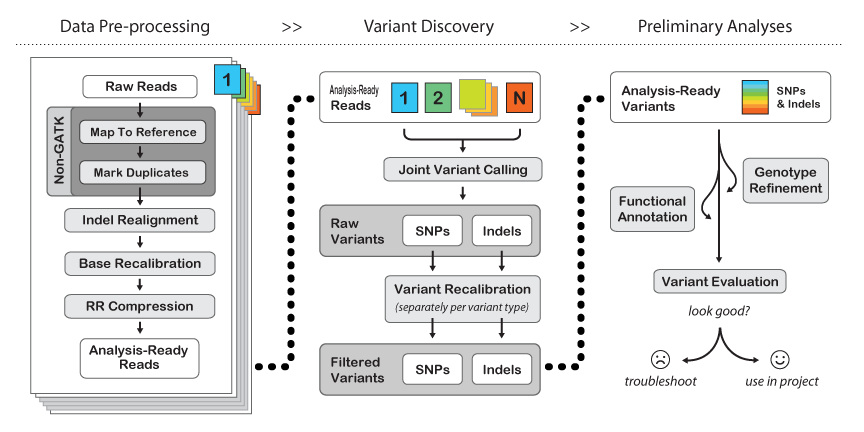
In brief, the key modifications made to the DNAseq Best Practices focus on handling splice junctions correctly, which involves specific mapping and pre-processing procedures, as well as some new functionality in the HaplotypeCaller. Here is a detailed overview:
- Tools: FastQC, MultiQC
- Steps:
- Run
FastQCon the raw FASTQ files to assess the quality of the sequencing reads. - Use
MultiQCto aggregate and visualize the results from multipleFastQCreports.
- Run
- Tools: Trimmomatic, Cutadapt
- Steps:
- Trim adapters and low-quality bases from the reads if necessary.
- Tools: STAR, HISAT2
- Steps:
- Align the RNA-Seq reads to the reference genome using
STARorHISAT2. - Generate a BAM file sorted by coordinates.
- Align the RNA-Seq reads to the reference genome using
- Tools: GATK (MarkDuplicates), Picard
- Steps:
- Use
GATK MarkDuplicatesorPicard MarkDuplicatesto identify and mark duplicate reads in the BAM file.
- Use
- Tools: GATK
- Steps:
- Use
GATK SplitNCigarReadsto split reads into exonic segments and hard-clip any overhanging portions. - Reassign mapping qualities from STAR (MAPQ=255) to a lower value (e.g., 60) using
SplitNCigarReads.
- Use
- Tools: GATK
- Steps:
- Perform Base Quality Score Recalibration (BQSR) using
GATK BaseRecalibrator. - Generate recalibrated BAM files.
- Perform Base Quality Score Recalibration (BQSR) using
- Tools: GATK HaplotypeCaller
- Steps:
- Run
GATK HaplotypeCallerwith the RNA-Seq-specific settings (-ERC GVCFmode recommended) to call variants.
- Run
- Steps:
- Combine GVCFs (if working with multiple samples) using
GATK CombineGVCFs. - Genotype GVCFs using
GATK GenotypeGVCFs.
- Combine GVCFs (if working with multiple samples) using
- Tools: GATK VariantFiltration
- Steps:
- Filter variants using
GATK VariantFiltrationwith appropriate RNA-Seq-specific filters. - For SNPs and indels, you may apply the following filters:
- SNPs:
QD < 2.0 || FS > 30.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 - Indels:
QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0
- SNPs:
- Filter variants using
- Tools: ANNOVAR, SnpEff, VEP
- Steps:
- Annotate the variants to determine their potential impact using
SnpEff,VEP, orANNOVAR.
- Annotate the variants to determine their potential impact using
- Tools: IGV (Integrative Genomics Viewer)
- Steps:
- Visualize the aligned reads and called variants in
IGVto ensure accuracy. - Interpret the variants in the context of the biological question being studied.
- Visualize the aligned reads and called variants in
This workflow should cover the essentials of RNA-Seq variant calling using GATK. Each tool has specific parameters that may need to be adjusted depending on the dataset and research question.