Publication-quality matplotlib figures for omics data, with two focuses:
-
omicsplot.heatmap— ComplexHeatmap-style heatmaps where the body is specified in absolute physical units (mm, cm, inch, pt). Surrounding panels (dendrograms, annotations, labels, colorbar, legend) grow outward without shrinking the body. Multi-panel composition with|(side-by-side) and/(stacked) keeps body positions exactly aligned across panels. Includes a chromosome-awaregenomic_heatmapfor binned signals (methylation, expression, copy number, accessibility). -
omicsplot.tracks— genome-wide track plots stacked vertically across per-chromosome subplot axes sized proportionally to chromosome lengths. Built-in track types: scatter, line segments, bar, step coverage, SV triangles. Trivially extensible by subclassing_BaseTrack.
Both submodules share a unit system inspired by R's grid::unit():
absolute (mm, cm, inch, pt), relative (npc), and flexible (null).
pip install omicsplotRequires Python ≥ 3.10. Depends on numpy, pandas, matplotlib, scipy, seaborn.
The snippets below show the API for each value proposition. Complete runnable examples — including synthetic data generation — are in docs/scripts/render_readme.py, which produces the figures shown here.
from omicsplot import unit
from omicsplot.heatmap import Heatmap, HeatmapAnnotation
hm = Heatmap(
df,
width=unit(120, 'mm'),
height=unit(50, 'mm'),
row_dendrogram_size=unit(12, 'mm'),
col_dendrogram_size=unit(10, 'mm'),
z_score=0, # row-wise normalization
cmap='RdBu_r',
center=0,
)
hm.add_top_annotation(HeatmapAnnotation(
group=sample_groups,
batch=sample_batch,
height=unit(3, 'mm'),
))
hm.draw()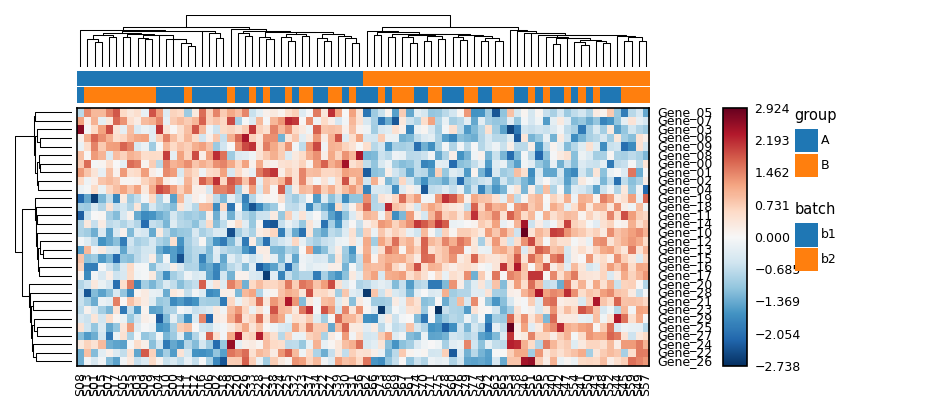
Use | for side-by-side and / for stacked. With share_row_order=True
(side-by-side) or share_col_order=True (stacked), bodies stay aligned
across panels.
hm_a = Heatmap(mat_a, width=unit(40, 'mm'), height=unit(45, 'mm'),
cmap='RdBu_r', center=0, name='Expression',
show_row_names=False)
hm_a.add_left_annotation(HeatmapAnnotation(
Group=groups, colors=group_colors, width=unit(4, 'mm'),
))
hm_b = Heatmap(mat_b, width=unit(25, 'mm'), height=unit(45, 'mm'),
cmap='YlOrRd', col_cluster=False, name='Copy number',
show_row_names=False)
(hm_a | hm_b).draw(share_row_order=True)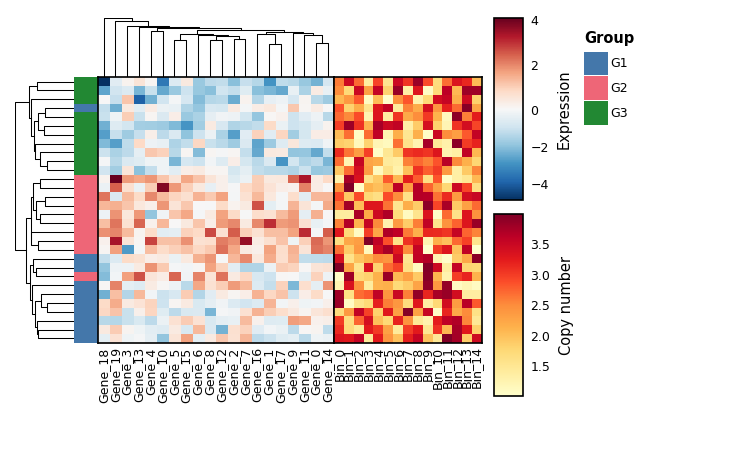
Compound layouts also work: (A | B) / C, A | B | C, etc.
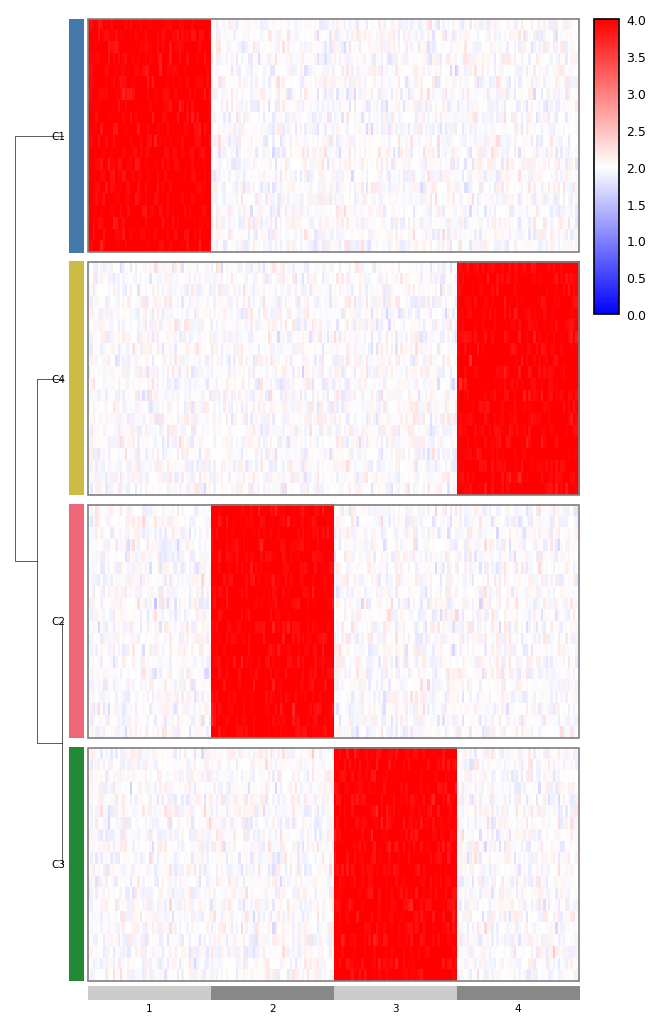
genomic_heatmap takes a long-form matrix with chrom/start/end
columns and arranges the columns into chromosome-proportional segments.
The optional cluster-of-clusters dendrogram (row_cluster_dendrogram)
shows relationships between groups, not individual rows.
from omicsplot.heatmap import genomic_heatmap
genomic_heatmap(
matrix, # columns: chrom, start, end, then per-cell
chrom_col='chrom',
cmap='bwr', vmin=0, vmax=4, center=2,
row_cluster=False,
show_row_names=False,
row_annotation={'Cluster': cluster_labels},
row_annotation_colors={'Cluster': palette},
row_gap=unit(2, 'mm'),
row_cluster_dendrogram=cluster_link, # leaves are groups
row_cluster_dendrogram_size=unit(10, 'mm'),
)
Each chromosome gets its own subplot axis sized proportionally to its
length. genome_range returns chromosome lengths for common builds.
from omicsplot import genome_range
from omicsplot.tracks import ScatterTrack, plot_tracks
gs = genome_range(genome='hg38')
scatter = ScatterTrack(
points, x_column='start', y_column='value',
ylabel='logR', ylim=(-1.5, 1.5),
s=1, alpha=0.4,
)
plot_tracks(scatter, gs, figsize=(10, 0.8))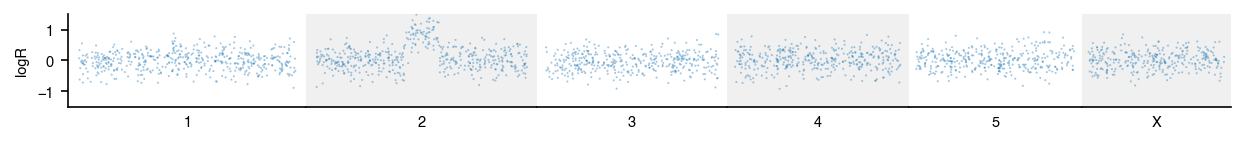
Alpha. API may change before 1.0.
MIT.