The GenoCore python version was developed by Seongmun Jeong and Jae-Yoon Kim.
This program is based on the GenoCore R version available at https://github.com/lovemun/Genocore.
Source code was written in Python language and supported on windows and linux platform.
git clone https://github.com/lovemun/GenoCore_Python
or
git clone https://github.com/JaeYoonKim72/GenoCore_Python
cd GenoCore_Python
The GenoCore consists of three submodules: VCFtoCSV, CoreSet, SelectVCF.
Basic usage : python run_genocore.py [ VCFtoCSV | CoreSet | SelectVCF ] [ Options ]
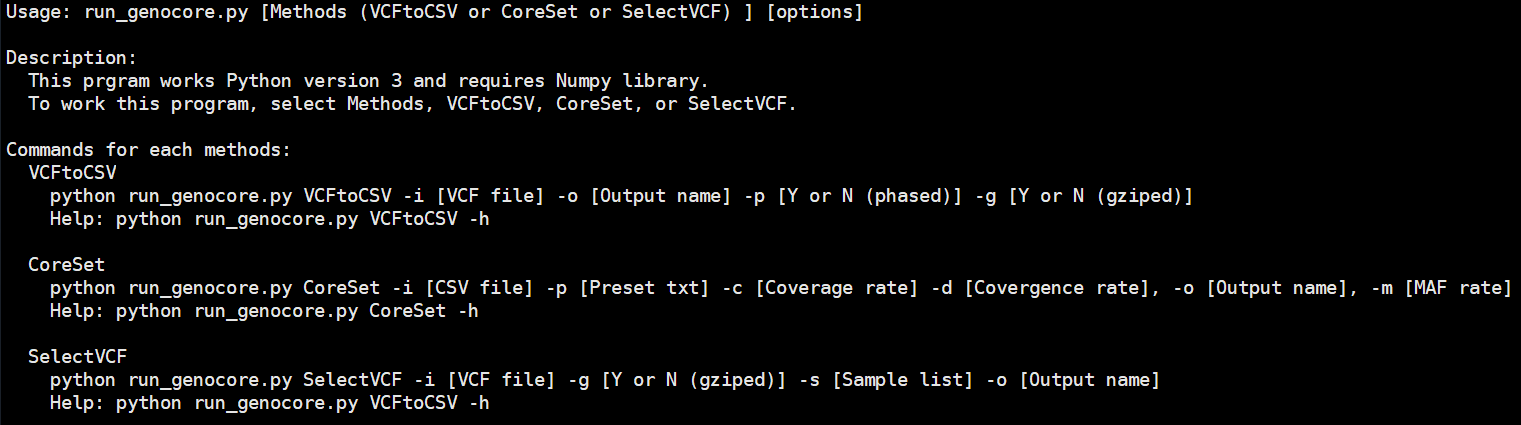
The "VCFtoCSV" submodule converts a VCF file into a CSV file for GenoCore. The output file is a CSV file for that VCF.
Usage: python run_genocore.py VCFtoCSV -i [VCF file] -o [Output name] -p [Y or N (phased)] -g [Y or N (gziped)]
Example: python run_genocore.py VCFtoCSV -i ExampleData/Test_420sample.vcf.gz \
-o ExampleData/Test_420sample.csv \
-p Y \
-g Y
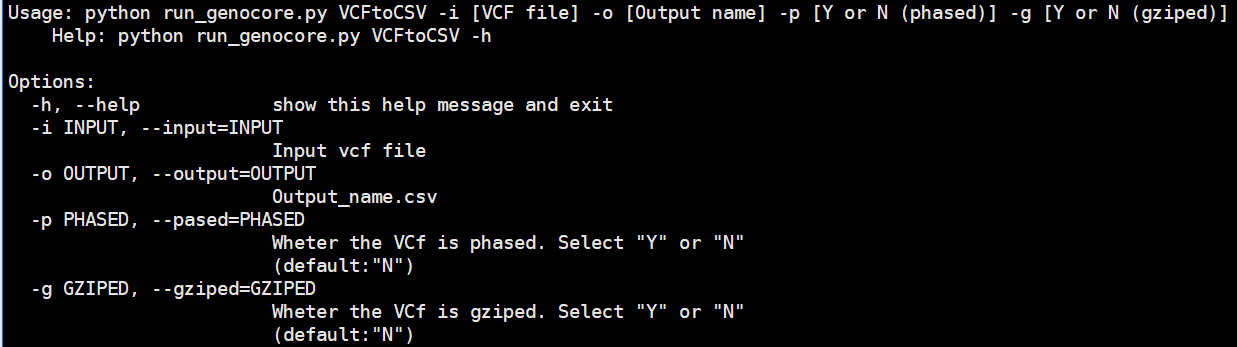
The "CoreSet" submodule is the main module and performs core sample extraction. The output files are core-sample-list, core-sample-csv and core-sample-coverage files, and removed-marker file.
Usage: python run_genocore.py CoreSet -i [CSV file] -p [Preset txt] -c [Coverage rate] -d [Covergence rate], -o [Output name], -m [MAF]
Example: python run_genocore.py CoreSet -i ExampleData/Test_420sample.csv \
-p ExampleData/Preset.txt \
-c 99 \
-d 0.001 \
-o ExampleData/TestCoreSet \
-m 0.05
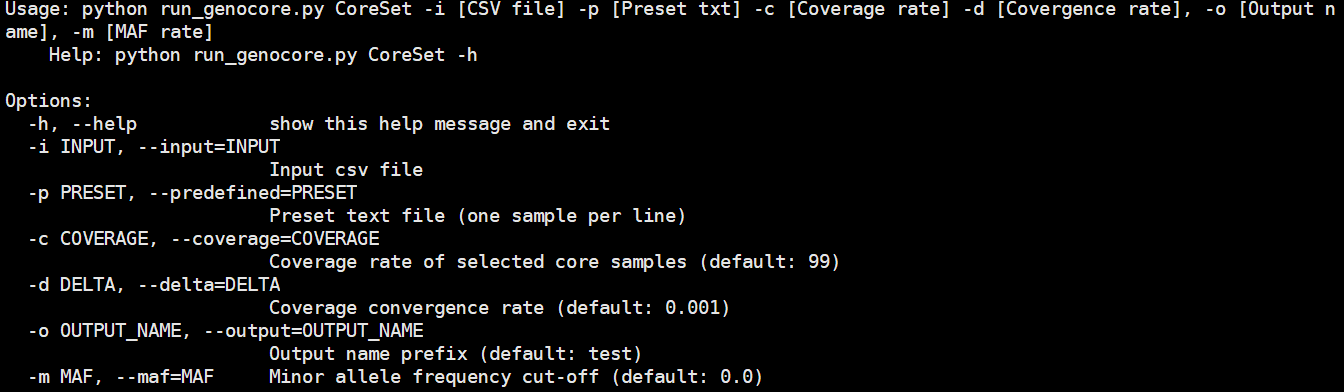
The "SelectVCF" submodule extract a core-set VCF file, using the core-sample-list file created by the "CoreSet" module and the input VCF file used by "VCFtoCSV". The output file is a VCF file for final core samples.
Usage: python run_genocore.py SelectVCF -i [VCF file] -g [Y or N (gziped)] -s [Sample list] -o [Output name]
Example: python run_genocore.py SelectVCF -i ExampleData/Test_420sample.vcf.gz \
-g Y \
-s ExampleData/TestCoreSet_CoreSample_list.txt \
-o ExampleData/TestCoreSet_CoreSample.vcf
The algorithm schematic of "CoreSet", the main submodule, is as follows.
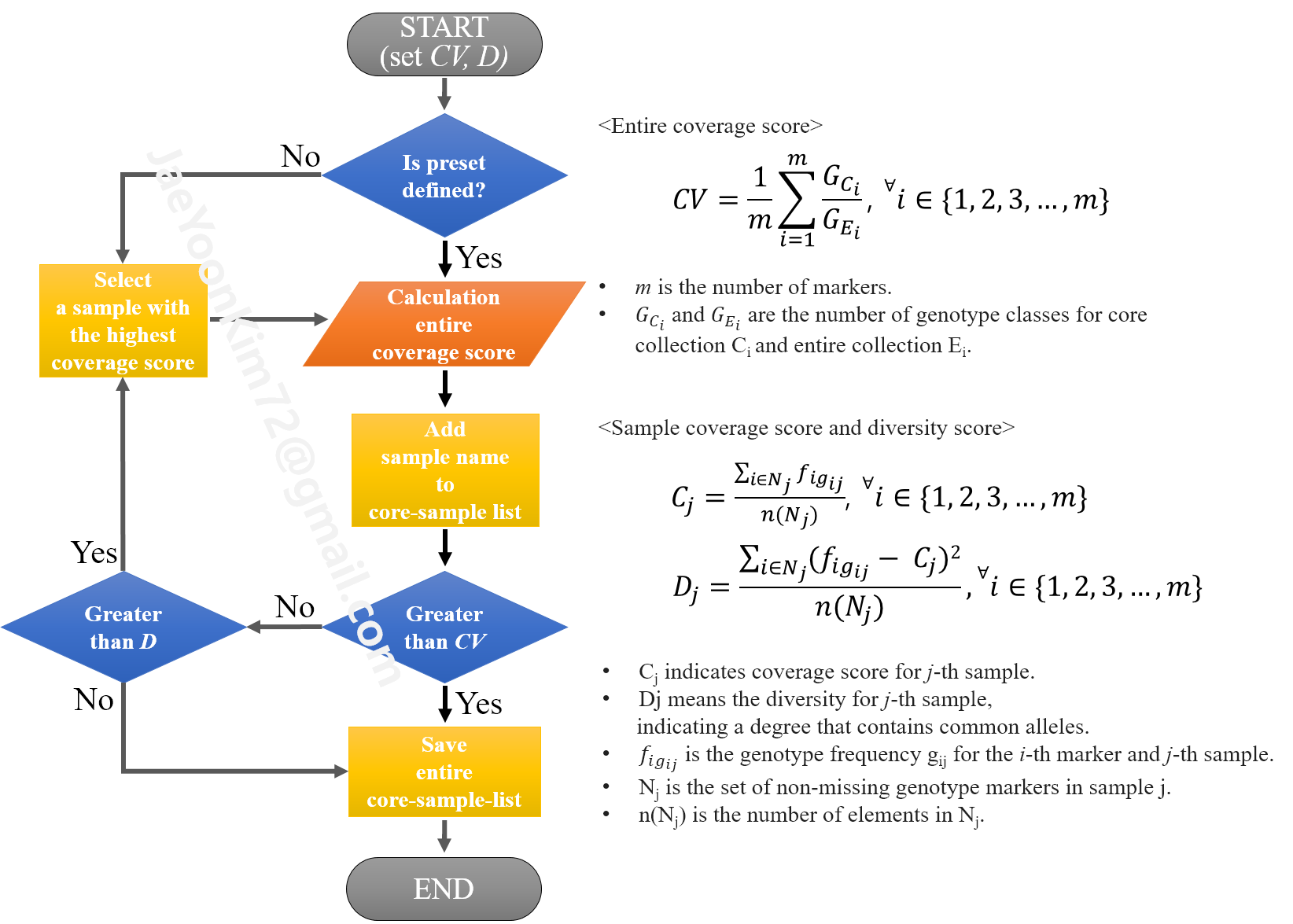
The GenoCroe python requires python 3.0 and numpy library. It also works in python 2.0, but python 3.0 is recommended for handling a large data set.
Jeong S, Kim JY, Jeong SC, Kang ST, Moon JK, et al. (2017) GenoCore: A simple and fast algorithm for core subset selection from large genotype datasets. PLOS ONE 12(7): e0181420. https://doi.org/10.1371/journal.pone.0181420