@kvkarandashev pointed out some problems to me for a simple benzene molecule
Training on random 3000 qm7 molecules gave a benzene molecule that was a bit off (see image). Especially the hydrogens were quite wrong. Increasing training set size improved the heavy atom coordinates but the hydrogens stayed out of plane.
An issue could be that aromaticity is not considered in the representations.
Theoretically, it isn't a problem to include this as long as there are parameters for it and it is represented in the graph via 1.5 bonds.
The problem is more on a technical side that getting a dataset and/or query that contains 1.5 bonds is more difficult.
RdKit has an aromatic bond type, but this requires some tweaking of xyz2mol. I'm also not sure whether xyz2mol actually detects aromaticity.
If someone queries a SMILES it would be easy to extract aromatic components, I see the problem more on the training data and parameter side.
Another way would be to augment the training data with resonance structures, but this would be more of a deep learning approach than KRR...
Any ideas/opinions @ferchault ?
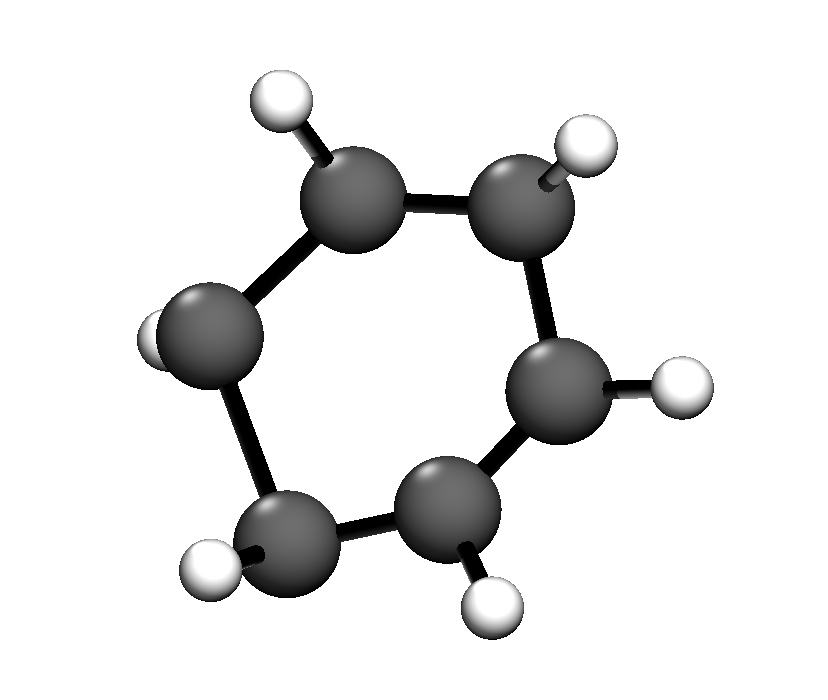
@kvkarandashev pointed out some problems to me for a simple benzene molecule
Training on random 3000 qm7 molecules gave a benzene molecule that was a bit off (see image). Especially the hydrogens were quite wrong. Increasing training set size improved the heavy atom coordinates but the hydrogens stayed out of plane.
An issue could be that aromaticity is not considered in the representations.
Theoretically, it isn't a problem to include this as long as there are parameters for it and it is represented in the graph via 1.5 bonds.
The problem is more on a technical side that getting a dataset and/or query that contains 1.5 bonds is more difficult.
RdKit has an aromatic bond type, but this requires some tweaking of xyz2mol. I'm also not sure whether xyz2mol actually detects aromaticity.
If someone queries a SMILES it would be easy to extract aromatic components, I see the problem more on the training data and parameter side.
Another way would be to augment the training data with resonance structures, but this would be more of a deep learning approach than KRR...
Any ideas/opinions @ferchault ?