Discrepancy between alignment and pileup summary #1835
Description
Are you using the latest version of samtools and HTSlib? If not, please specify.
% samtools --version
samtools 1.17
Please describe your environment.
- OS: Darwin 22.3.0
- machine architecture: x86_64
- compiler:
Apple clang version 14.0.3 (clang-1403.0.22.14.1)
Target: x86_64-apple-darwin22.3.0
Thread model: posix
InstalledDir: /Library/Developer/CommandLineTools/usr/bin
Please specify the steps taken to generate the issue, the command you are running and the relevant output.
I am finding a significant discrepancy between a pileup summary (output of samtools mpileup) and the original alignment. I am not sure if this is an error on my part or on the part of mpileup.
I am trying to generate a pileup summary for nanopore sequencing reads that were used to generate a consensus sequence. The idea is to determine specific base pairs of the consensus that are more questionable than others by evaluating their quality score and number of errors. Using my initial test data, I have the following consensus sequence, generated from the following binned read sequences.
consensus00.fastq.tar.gz
targetSequenceBin00000.fastq.tar.gz
I unfortunately cannot use the alignment originally used to generate the consensus sequence - it was made using mafft, which does not produce a SAM or BAM file. I used the following alignment code to generate a SAM file by aligning reads to the consensus.
minimap2 -a consensus00.fastq targetSequenceBin00000.fastq > 00.sam
which makes:
00.sam.tar.gz
This resulting SAM file seems to work - when I visualize the alignment the depiction forms with no errors. Note that using Tablet demonstrates that 217 sequences mapped to the initial 'T' of the consensus sequence:

I ran this program through several samtools commands to prep it for samtools mpileup, namely the following:
samtools view 00.sam -b -o 00.bam
samtools sort 00.bam -o 00_sort.bam
samtools mpileup 00_sort.bam -f consensus00.fastq -o 00_full.tsv -s -O
Doing so I successfully get a pileup summary:
00_full.tsv.tar.gz
The problem is, when you look at the first few lines of this pileup summary, you get some discrepancies with the original SAM file. Where the first 'T' had 217 reads mapped at that location, the pileup summary only has 40.
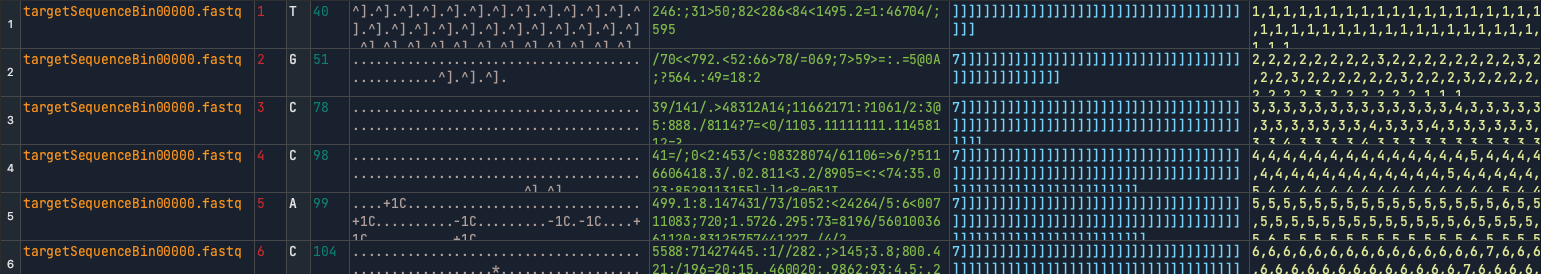
I don't know what is causing the discrepancy between the original alignment and the pileup summary. It may be this is an element of the pileup summary I am missing, or it may be a bug, or it may be the alignment was changed in the processing step leading up to the pileup summary. Are you aware of what may have caused this discrepancy?