Hi!
I have a problem with the query function. I used it extract R2 previously, and it seems to works fine, all the variants are there. But when I try to extract "GENOTYPED" from the INFO-field, and there are trouble with the output. "GENOTYPED" is only there for the genotyped markers, and using query to extract the field seems to work fine when having a quick look; the genotyped variants will have a "1" and the rest will have a ".".
But when I look closer, there seem to be a problem with bcftools suddenly jumping from one variant, to another one, and not the next one, in the middle of the line.
It is probably easier explained with a picture of the output.
This is the outputfile
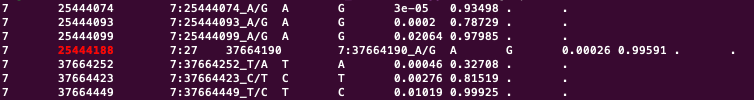
and this is the original vcf-fila
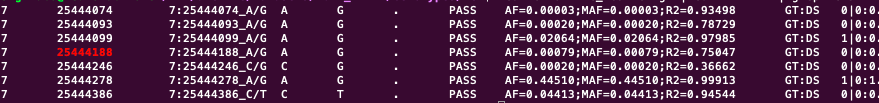
As you can see, in the outputfile, while writing variant on position 25444188, it suddenly jumps to variant 37664252, in the middle of the line. This happens in all the chromosomes. It also happens in different datasets.
I cannot see anything wrong with the vcf-file, and it seems to happen with random variants (i run the query twice, and the mistake was seen at different variants).
Since it only happens when trying to include an INFO-field which is not there for all variants, I am guessing it is related to this, even if it does not seem to be a mistake that occurs at those variants.
(I ended up just grep'ing out the variants with the GENOTYPE-flag but thought I´d post this issue anyway)
Thanks
Hi!
I have a problem with the query function. I used it extract R2 previously, and it seems to works fine, all the variants are there. But when I try to extract "GENOTYPED" from the INFO-field, and there are trouble with the output. "GENOTYPED" is only there for the genotyped markers, and using query to extract the field seems to work fine when having a quick look; the genotyped variants will have a "1" and the rest will have a ".".
But when I look closer, there seem to be a problem with bcftools suddenly jumping from one variant, to another one, and not the next one, in the middle of the line.
It is probably easier explained with a picture of the output.
This is the outputfile
and this is the original vcf-fila

As you can see, in the outputfile, while writing variant on position 25444188, it suddenly jumps to variant 37664252, in the middle of the line. This happens in all the chromosomes. It also happens in different datasets.
I cannot see anything wrong with the vcf-file, and it seems to happen with random variants (i run the query twice, and the mistake was seen at different variants).
Since it only happens when trying to include an INFO-field which is not there for all variants, I am guessing it is related to this, even if it does not seem to be a mistake that occurs at those variants.
(I ended up just grep'ing out the variants with the GENOTYPE-flag but thought I´d post this issue anyway)
Thanks