MetaPhlAn 4.1
MetaPhlAn is a tool for profiling the composition of microbial communities from metagenomic shotgun sequencing data.
- For information about installation and pre-requisites for MetaPhlAn 4.1, please see the manual page.
- Please direct science and software support questions to the MetaPhlAn bioBakery forum topic.
- Legacy tutorials for previous MetaPhlAn versions are also available:
If you're using this tutorial as part of a workshop, or if you'd like to think about the algorithm and implementation details a bit, you'll occasionally see "discussion questions" at the end of tutorial sections, formatted like this:
- What is your quest?
- What is your favorite color?
- 1. Creating taxonomic profiles
- 2. Visualizing taxonomic profiles
- 3. MetaPhlAn viral module tutorial
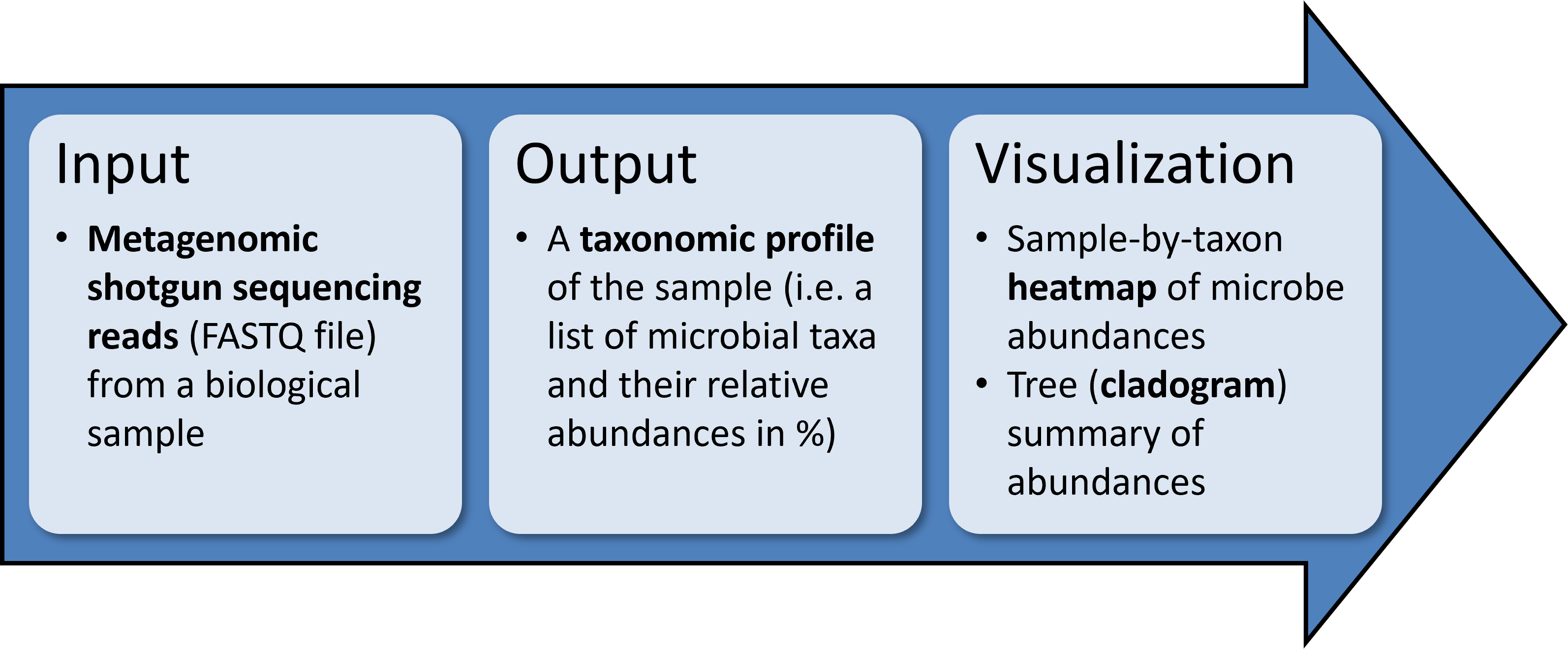
MetaPhlAn accepts as input short reads from a single shotgun metagenomic sequencing experiment and outputs the list of detected microbes and their relative abundances.
MetaPhlAn accepts metagenomic sequence information in several formats, including fastq, fasta, sam, and bowtie2out (a MetaPhlAn-specific mapping summary).
To see all the input formats (and other arguments), type metaphlan -h | less. Use the arrow key to move up and down. Type q to quit back to the command prompt.
usage: metaphlan --input_type {fastq,fasta,bowtie2out,sam} [--force]
[--bowtie2db METAPHLAN_BOWTIE2_DB] [-x INDEX]
[--bt2_ps BowTie2 presets] [--bowtie2_exe BOWTIE2_EXE]
[--bowtie2_build BOWTIE2_BUILD] [--bowtie2out FILE_NAME]
[--min_mapq_val MIN_MAPQ_VAL] [--no_map] [--tmp_dir]
[--tax_lev TAXONOMIC_LEVEL] [--min_cu_len]
[--min_alignment_len] [--add_viruses] [--ignore_eukaryotes]
[--ignore_bacteria] [--ignore_archaea] [--ignore_ksgbs]
[--ignore_usgbs] [--stat_q] [--perc_nonzero]
[--ignore_markers IGNORE_MARKERS] [--avoid_disqm] [--stat]
[-t ANALYSIS TYPE] [--nreads NUMBER_OF_READS]
[--pres_th PRESENCE_THRESHOLD] [--clade] [--min_ab]
[--profile_vsc] [--vsc_out VSC_OUT]
[--vsc_breadth VSC_BREADTH] [-o output file]
[--sample_id_key name] [--use_group_representative]
[--sample_id value] [-s sam_output_file] [--legacy-output]
[--CAMI_format_output] [--unclassified_estimation] [--mpa3]
[--biom biom_output] [--mdelim mdelim] [--nproc N]
[--subsampling SUBSAMPLING] [--mapping_subsampling]
[--subsampling_seed SUBSAMPLING_SEED]
[--subsampling_output SUBSAMPLING_OUTPUT] [--install]
[--offline] [--force_download] [--read_min_len READ_MIN_LEN]
[-v] [-h]
[INPUT_FILE] [OUTPUT_FILE]
DESCRIPTION
MetaPhlAn version 4.1.0 (23 Aug 2023):
METAgenomic PHyLogenetic ANalysis for metagenomic taxonomic profiling.
AUTHORS: Aitor Blanco-Miguez (aitor.blancomiguez@unitn.it), Francesco Beghini (francesco.beghini@unitn.it), Moreno Zolfo (moreno.zolfo@unitn.it), Nicola Segata (nicola.segata@unitn.it), Duy Tin Truong, Francesco Asnicar (f.asnicar@unitn.it)
- Which command line arguments are required?
- Which optional command line arguments seem most important or most commonly used?
For the purpose of this tutorial, we will use the following set of six human metagenomes from the Human Microbiome Project (HMP) which have been downsampled for rapid analysis:
- SRS014476-Supragingival_plaque.fasta.gz
- SRS014494-Posterior_fornix.fasta.gz
- SRS014459-Stool.fasta.gz
- SRS014464-Anterior_nares.fasta.gz
- SRS014470-Tongue_dorsum.fasta.gz
- SRS014472-Buccal_mucosa.fasta.gz
The original files, and many others, can be downloaded from the HMP DACC.
- If you are running this tutorial as a part of short course on a bioBakery cloud instance, the input files have been pre-downloaded. Open a terminal and enter:
cd Tutorials/metaphlan4
ls input
- Copy the input files into the current working directory
cp input/*.fasta.gz .
-
Use the
lscommand to check if the files have been correctly copied/downloaded. -
We are now ready to run MetaPhlAn. Please proceed to the next section of the tutorial.
-
IF you're running this tutorial locally (your PC/laptop), make a new folder in your current working directory with
mkdirand access it withcd. Use this folder for all MetaPhlAn analyses in this tutorial.
mkdir metaphlan_analysis
cd metaphlan_analysis
- Click on the links above to download each HMP file. NOTE that if you are using the Google Chrome browser, it may automatically decompress (gunzip) the gzip-ed files. The files should download to your default
Downloads/folder location. Copy the files fromDownloads/to yourmetaphlan_analysis/folder:
cp ~/Downloads/SRS*.fasta.gz .
- Alternatively, you can use the
curlorwgetprograms on MacOS / Linux to download each file directly into the folder you created. Confirm that you are working in themetaphlan_analysis/folder withpwd. Right click on each link, choose "Copy Link Address" (or equivalent), and in a terminal type:
curl -LO $LINK (OR) wget $LINK
-
Where
$LINKis the link you copied to your clipboard. -
Use the
lscommand to verify that the files have been correctly copied/downloaded. Each should be about 700 Kb. We are now ready to run MetaPhlAn.
- If you're familiar with the command line and file structure, does it matter where you place MetaPhlAn's input files, or where you run it from?
- What about these input files might make them particularly appropriate for a short demonstration?
Here is a minimal command for profiling a single metagenome with MetaPhlAn (using the first downsampled HMP file described above as an input):
metaphlan SRS014476-Supragingival_plaque.fasta.gz --input_type fasta > SRS014476-Supragingival_plaque_profile.txt
(NOTE: If this is your first time running MetaPhlAn, the first step in this process involves MetaPhlAn downloading, decompressing, and indexing its marker gene database. This process may take ~30 minutes - depending in part on your download speed - but only needs to be performed once.)
Running MetaPhlAn following the example in the prior section will create two output files. Check what files have been created with ls.
SRS014476-Supragingival_plaque.fasta.gz.bowtie2out.txt
This file contains a reduced representation of the (intermediate) mapping results of your sequencing reads to MetaPhlAn's marker gene sequences. Alignments are listed one-per-line in tab-separated columns of Read ID and Marker Gene ID. Inspect the file by executing:
less -S SRS014476-Supragingival_plaque.fasta.gz.bowtie2out.txt
Which yields:
HWUSI-EAS1568_102539179:1:100:10001:9884/1__1.14 VDB|0021-0089-0-001A|M364-c1468-c0-c0
HWUSI-EAS1568_102539179:1:100:10005:18658/1__1.36 UniRef90_E0DG58|1__8|SGB17007
HWUSI-EAS1568_102539179:1:100:10007:7282/1__1.50 UniRef90_P44049|1__7|SGB9649
HWUSI-EAS1568_102539179:1:100:10008:17064/1__1.53 UniRef90_E0DI50|1__4|SGB17007
HWUSI-EAS1568_102539179:1:100:10011:6241/1__1.79 UniClust90_MPCHLGFL01100|1__5|SGB15899
HWUSI-EAS1568_102539179:1:100:10013:1452/1__1.89 VDB|0003-004B-0-0000|M1312-c1887-c0-c4
HWUSI-EAS1568_102539179:1:100:10013:7741/1__1.92 UniClust90_ENKOMPNM00366|1__8|SGB19880
HWUSI-EAS1568_102539179:1:100:10024:13157/1__1.141 UniRef90_E0DFH6|1__3|SGB17007
HWUSI-EAS1568_102539179:1:100:10025:5272/1__1.159 UniRef90_A0A3S5F533|1__7|SGB17007
HWUSI-EAS1568_102539179:1:100:10035:9129/1__1.208 UniClust90_FHIIFBMB00432|1__12|SGB69135
...
- What do the parts of the read identifiers in the 1st column indicate?
- What do the parts of the gene identifiers in the 2nd column indicate?
SRS014476-Supragingival_plaque_profile.txt
This file contains the final computed taxon abundances. Taxon abundances are listed one clade per line, tab-separated from the clade's relative abundance in %:
less -S SRS014476-Supragingival_plaque_profile.txt
Which yields:
[1] # DATABASE VERSION
[2] # COMMAND EXECUTED
[3] # 19048 reads processed
[4] # SampleID Metaphlan_Analysis
[5] # clade_name NCBI_tax_id relative_abundance additional_species
k__Bacteria 2 100.0
k__Bacteria|p__Actinobacteria 2|201174 85.43048
k__Bacteria|p__Firmicutes 2|1239 14.56952
k__Bacteria|p__Actinobacteria|c__Actinomycetia 2|201174|1760 85.43048
k__Bacteria|p__Firmicutes|c__Bacilli 2|1239|91061 14.56952
...
The file has a five line header (lines beginning with #). We've added [i]s (line numbers) to the header text above to help call out their roles more clearly.
-
[1]: Indicates the version of the marker gene database that MetaPhlAn used in this run. There are currently ~1.1M unique clade-specific marker genes identified from ~100k reference genomes (~99,500 bacterial and archaeal and ~500 eukaryotic) included in the database. -
[2]: Provides a copy of the command that was run to produce this profile. This includes the path to the MetaPhlAn executable, the input file that was analyzed, and any custom parameters that were configured. -
[3]: Indicates the number of sample reads processed. -
[4]: Lists your sample name. -
[5]: Provides column headers from the profile data that follows.
More specifically, [5] includes four headers to be familiar with:
-
clade_name: The taxonomic lineage of the taxon reported on this line, ranging from Kingdom (e.g. Bacteria/Archaea) to species genome bin (SGB). Taxon names are prefixed to help indicate their rank:Kingdom: k__,Phylum: p__,Class: c__,Order: o__,Family: f__,Genus: g__,Species: s__, andSGB: t__. We'll examine these more closely below. -
NCBI_tax_id: The NCBI-equivalent taxon IDs of the named taxa fromclade_name. -
relative_abundance: The taxon's relative abundance in %. Since typical shotgun-sequencing-based taxonomic profile is relative (i.e. it does not provide absolute cell counts), clades are hierarchically summed. Each taxonomic level will sum to 100%. That is, the sum of all kingdom-level clades is 100%, the sum of all phylum-level clades is 100%, and so forth. -
additional_species: Additional species names for cases where the metagenome profile contains clades that represent multiple species. The species listed in column 1 is the representative species in such cases.
Let's use grep to zoom in on a more specific example, the taxonomic lineage of the first species in the file:
grep s__ SRS014476-Supragingival_plaque_profile.txt | head -1 | cut -f1 | sed 's/|/\n/g'
(Command explained: grep s__ isolates species-resolved lineages; which are "piped" with | to head -1, which extracts just the first species; cut -f1 extracts just the first column value - taxonomy; and the sed command replaces the |s in the lineage with newlines.)
This yields the taxonomy of the microbe C. matruchotii:
k__Bacteria
p__Actinobacteria
c__Actinomycetia
o__Corynebacteriales
f__Corynebacteriaceae
g__Corynebacterium
s__Corynebacterium_matruchotii
As an additional experiment, let's verify that the sum of all taxonomic orders' abundances is 100%:
grep o__ SRS014476-Supragingival_plaque_profile.txt | grep -v f__ | cut -f1,3
(Command explained: grep o__ isolates order-resolved lineages while grep -v f__ removes family-resolved lineages, leaving orders as the most specific taxa; cut -f 1,3 then isolates the taxonomy column and the relative abundance column.)
Which yields:
k__Bacteria|p__Actinobacteria|c__Actinomycetia|o__Corynebacteriales 53.75141
k__Bacteria|p__Actinobacteria|c__Actinomycetia|o__Micrococcales 31.67907
k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales 14.56952
This looks good by visual inspection! We can modify the command above to verify the sum at other taxonomic levels, e.g. species:
grep s__ SRS014476-Supragingival_plaque_profile.txt | grep -v t__ | cut -f1,3 | sed "s/.*|//g"
(Command explained: This command adds an additional call to sed to remove the taxonomy up to the species prefix, which helps to make the output a little easier to read.)
Which yields:
s__Corynebacterium_matruchotii 53.75141
s__Rothia_SGB49305 31.67907
s__Streptococcus_sanguinis 14.56952
-
Since the per-clade abundance output file is already normalized, you never need to sum-normalize these relative abundances. However, if you tried to, what would the sum of all clades' relative abundances be?
-
Why do the four species extracted above have the same relative abundances as the four orders extracted with the previous command? (Hint: remove the
sedcommand from the species version to see their full taxonomic lineages.)
When re-analyzing a sample (e.g. using different MetaPhlAn options), it is preferable to start from the sample's .bowtie2out.txt file. This allows you to skip the time-consuming step of mapping the sample's reads to the marker gene database, making the re-analysis much faster.
Let's explore this option by deleting the profile we created above and then regenerating it, this time starting from the .bowtie2out.txt file.
rm -f SRS014476-Supragingival_plaque_profile.txt
Note that we will need to change the --input_type argument from our original command since we are not starting from raw reads:
metaphlan SRS014476-Supragingival_plaque.fasta.gz.bowtie2out.txt --input_type bowtie2out > SRS014476-Supragingival_plaque_profile.txt
This command should finish notably faster than the original run (which was already fast). Verify that the regenerated profile matches the one produced directly from the sample's fastq file (e.g. by extracting and examining species-level abundances using the grep commands introduced in the previous section).
MetaPhlAn can take advantage of multiple cores using the nproc flag. Let's profile one of the other HMP samples, again starting from raw reads, this time using 4 cores during the read-mapping stage:
metaphlan SRS014459-Stool.fasta.gz --input_type fasta --nproc 4 > SRS014459-Stool_profile.txt
(Note: nproc is used by bowtie2 which processes 10K reads per second per thread. Since we have a very small number of reads in this demo, the difference in speed up is negligible.)
Each MetaPhlAn execution processes exactly one sample, but the resulting single-sample analyses can easily be combined into an abundance table spanning multiple samples. Let's finish the last four samples from the input files tutorial section:
metaphlan SRS014464-Anterior_nares.fasta.gz --input_type fasta --nproc 4 > SRS014464-Anterior_nares_profile.txt
metaphlan SRS014470-Tongue_dorsum.fasta.gz --input_type fasta --nproc 4 > SRS014470-Tongue_dorsum_profile.txt
metaphlan SRS014472-Buccal_mucosa.fasta.gz --input_type fasta --nproc 4 > SRS014472-Buccal_mucosa_profile.txt
metaphlan SRS014494-Posterior_fornix.fasta.gz --input_type fasta --nproc 4 > SRS014494-Posterior_fornix_profile.txt
Alternatively, if you are familiar with shell syntax, you can loop over all input files (make sure you have deleted previously generated Bowtie 2 output files to prevent errors):
for i in SRS*.fasta.gz; do metaphlan $i --input_type fasta --nproc 4 > ${i%.fasta.gz}_profile.txt; done
Either way, you will now have a complete set of six profile output files and six intermediate mapping files. If you'd like to skip this step to
speed things up, the 12 demo file outputs can be downloaded from the following links (right-click on the link and pick "Save Link as..." or click on the link and then right-click on the preview page and select "Save Page as...", or copy the URL to download on a server).
-
If you are running this tutorial as part of a short course, the Bowtie 2 output files and profile files are in the
output/folder. To copy them to the current directory execute:
cp output/*_profile.txt .
- SRS014459-Stool_profile.txt
- SRS014464-Anterior_nares_profile.txt
- SRS014470-Tongue_dorsum_profile.txt
- SRS014472-Buccal_mucosa_profile.txt
- SRS014476-Supragingival_plaque_profile.txt
- SRS014494-Posterior_fornix_profile.txt
- SRS014459-Stool.fasta.gz.bowtie2out.txt
- SRS014464-Anterior_nares.fasta.gz.bowtie2out.txt
- SRS014470-Tongue_dorsum.fasta.gz.bowtie2out.txt
- SRS014472-Buccal_mucosa.fasta.gz.bowtie2out.txt
- SRS014476-Supragingival_plaque.fasta.gz.bowtie2out.txt
- SRS014494-Posterior_fornix.fasta.gz.bowtie2out.txt
Finally, the MetaPhlAn distribution includes a utility script that will create a single tab-delimited table from a set of sample-specific abundance profiles (isolating the sample names, feature taxonomies, and relative abundances):
merge_metaphlan_tables.py *_profile.txt > merged_abundance_table.txt
You can also download the merged table from here:
This table can be opened as a spreadsheet (using a program like Excel), any gene expression analysis program, less (example below), or visualized graphically as per subsequent tutorial sections:
less -S merged_abundance_table.txt
The first few lines look like:
#mpa_vJun23_CHOCOPhlAnSGB_202307
clade_name SRS014459-Stool SRS014464-Anterior_nares SRS014470-Tongue_dorsum SRS014472-Buccal_mucosa SRS014476-Supragingival_plaque SRS014494-Posterior_fornix
UNCLASSIFIED 100.0 0.0 0.0 0.0 0.0 0.0
k__Bacteria 0.0 100.0 100.0 100.0 100.0 100.0
k__Bacteria|p__Proteobacteria 0.0 81.20128 0.0 14.7449 0.0 0.0
k__Bacteria|p__Actinobacteria 0.0 18.79872 0.0 0.0 85.43048 0.0
k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria 0.0 81.20128 0.0 14.7449 0.0 0.0
k__Bacteria|p__Actinobacteria|c__Actinomycetia 0.0 18.79872 0.0 0.0 85.43048 0.0
k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Moraxellales 0.0 81.20128 0.0 0.0 0.0 0.0
k__Bacteria|p__Actinobacteria|c__Actinomycetia|o__Corynebacteriales 0.0 18.79872 0.0 0.0 53.75141 0.0
-
How might you convert these relative abundance measures to pseudo-RPKs that are sensitive to each sample's read depth?
-
In what important ways does analysis of a relative abundance table differ from that of a gene expression (microarray or RNA-seq) transcript table? In what ways are they similar?
-
Under what circumstances is this tab-delimited text data format particularly efficient or inefficient? Is this likely to be a problem for species-level taxonomic profiles?
A heatmap is one way to visualize tabular abundance results such as those from MetaPhlAn. The plotting tool we'll use here, hclust2, is a convenience script that can show any, some, or all of the microbes or samples in a MetaPhlAn table. In this tutorial we will plot the heatmap for all of the samples.
If you're using this tutorial from the bioBakery VM (locally or on the cloud), hclust2 is already installed! Otherwise, you can install hclust2 and other bioBakery tools automatically with Conda. :
conda install -c biobakery hclust2
This will install hclust2 and all of its dependencies.
Run the following command to create a species only abundance table, providing the abundance table (merged_abundance_table.txt) created in prior tutorial steps as the first file argument in the chain:
grep -E "s__|SRS" merged_abundance_table.txt \
| grep -v "t__" \
| sed "s/^.*|//g" \
| sed "s/SRS[0-9]*-//g" \
> merged_abundance_table_species.txt
(Command explained: The initial grep command is isolating the header row, which matches SRS, or any species-resolved row containing s__; the second grep command then excludes rows that are also resolved to the SGB level, matching t__; the first sed command removes everything up to the s__ prefix from the species' taxonomies; the second sed command simplifies the sample names.)
The new abundance table (merged_abundance_table_species.txt) will contain only the species abundances with just the species names as the row headers (instead of their full taxonomy) and just the body site names as the column headers (removing the specific SRS sample numbers).
The first few lines of the file will look like:
clade_name Stool Anterior_nares Tongue_dorsum Buccal_mucosa Supragingival_plaque Posterior_fornix
s__Moraxella_nonliquefaciens 0.0 81.20128 0.0 0.0 0.0 0.0
s__Corynebacterium_pseudodiphtheriticum 0.0 18.79872 0.0 0.0 0.0 0.0
s__Veillonella_dispar 0.0 0.0 74.02531 0.0 0.0 0.0
s__Prevotella_histicola 0.0 0.0 25.97469 0.0 0.0 0.0
s__Streptococcus_mitis 0.0 0.0 0.0 85.2551 0.0 0.0
s__Haemophilus_haemolyticus 0.0 0.0 0.0 14.7449 0.0 0.0
s__Corynebacterium_matruchotii 0.0 0.0 0.0 0.0 53.75141 0.0
s__Rothia_SGB49305 0.0 0.0 0.0 0.0 31.67907 0.0
s__Streptococcus_sanguinis 0.0 0.0 0.0 0.0 14.56952 0.0
s__Lactobacillus_crispatus 0.0 0.0 0.0 0.0 0.0 84.0836
s__Lactobacillus_iners 0.0 0.0 0.0 0.0 0.0 14.40927
s__Lactobacillus_jensenii 0.0 0.0 0.0 0.0 0.0 1.50713
- Why is it useful to visualize (and particularly to cluster) only the "tips" of the taxonomic tree?
Next generate the species only heatmap by running the following command:
hclust2.py \
-i merged_abundance_table_species.txt \
-o metaphlan4_abundance_heatmap_species.png \
--f_dist_f braycurtis \
--s_dist_f braycurtis \
--cell_aspect_ratio 0.5 \
--log_scale \
--flabel_size 10 --slabel_size 10 \
--max_flabel_len 100 --max_slabel_len 100 \
--minv 0.1 \
--dpi 300
We have only 12 microbes in this demo file but typically, for ease of viewing, one can show the top 25 species using the --ftop 25 argument. This command 1) uses Bray-Curtis as the distance measure both between samples (s) and between features (f) (i.e. species), 2) sets the ratio between the width/height of cells to 0.5, 3) uses a log scale for assigning heatmap colors, 4) sets the sample and feature label size to 10, 5) sets the max sample and feature label length to 100, 6) selects the minimum value to display as 0.1, and 7) selects an image resolution of 300 (in that order!). You can learn more about these options by executing hclust2.py --help.
Open the resulting heatmap (metaphlan4_abundance_heatmap_species.png) to take a look. If you generated it on your local computer, just double click. If you're using a server with just a terminal interface, you might have to transfer the file locally first using a tool like scp. If you're using a server with a graphical interface, you can open the file using a command like see metaphlan4_abundance_heatmap_species.png). Using any of these methods, the results should look like:

Notice that due to the very large differences between body site communities in the human microbiome, we can still easily see site-specific species despite the small demonstration input files (each is subsampled to <20k reads for efficiency). However, no species were detected for the stool sample in this example due to the low number of reads used.
- Which microbes are most abundant at each body site in these demonstration data?
- Under what circumstances is log-scaling the heatmap abundance colors good? When might it be bad (i.e. visually deceptive)?
(Coming soon! This section of the tutorial is being updated for compatiblility with MetaPhlAn 4's updated SGB taxonomy. For a similar analysis, check out the cladograph construction section of the MetaPhlAn 3 tutorial.)
You can download this tutorial dataset containing two subsampled FASTQ files: Example Dataset
To profile a sample, simply add the --profile_vsc option to the standard command.
For example, the following command profiles SRS014287_HMP_2012.fastq.gz using 4 cpus (it can take a few minutes).
# Read SRS014287_HMP_2012.fastq.gz and profile VSCs
zcat SRS014287_HMP_2012.fastq.gz | metaphlan \
--input_type fastq --profile_vsc \
--nproc 4 -o SRS014287_HMP_2012.profile.txt \
--vsc_out SRS014287_HMP_2012.vsc.txt;
The breadth and depth of coverage of each group will be saved in the path provided with --vsc_out (in this case: SRS014287_HMP_2012.vsc.txt)
| M-Group Cluster | genomeName | len | breadth of coverage | depth of coverage mean | depth of coverage median | M-Group-Type [k|u] | First Genome in Cluster | Other Genomes |
|---|---|---|---|---|---|---|---|---|
| M1102 | VDB|0028-001B-0-0004|M1102-c72-c0-c73 | 1569 | 1 | 229.39 | 243 | kVSG | NC_024711_Uncultured_crAssphage | NC_0247111_Uncultured_crAssphage |
| M738 | VDB|0028-005D-0-0008|M738-c70-c0-c12 | 1658 | 1 | 248.07 | 266 | uVSG | - | - |
| M616 | VDB|0021-00AB-0-0036|M616-c2556-c1-c0 | 2819 | 1 | 5.16 | 3 | uVSG | - | - |
| M697 | VDB|0030-0055-0-0000|M697-c3861-c6-c10 | 5920 | 0.99 | 25.79 | 30 | uVSG | - | - |
| M1081 | VDB|002C-001A-0-0004|M1081-c3793-c0-c0 | 14046 | 0.87 | 6.75 | 7 | uVSG | - | - |
Use the --vsc_breadth option to change the reporting filter on the breadth of coverage (default is 0.75, meaning 75%). Groups with a breadth of coverage below this threshold will not be reported.
Use the merge_vsc_tables.py command to merge individual profiles into one table:
# Merge multiple tables
merge_vsc_tables.py -o OUT.vsc.txt *_vsc.txt
A grouped table will be saved in OUT.vsc.txt. The default value is the breadth of coverage, but you can specify other values with the -g option.
| M-Group/Cluster | M-Group-Type [k|u] | First Genome in Cluster | … | SRR8653074_LiangG_2020.profile_vsc | SRS014287_HMP_2012.profile_vsc |
|---|---|---|---|---|---|
| M1081 | uVSG | - | … | 0.0000 | 0.8701 |
| M1102 | kVSG | NC_024711_Uncultured_crAssphage | … | 0.0000 | 1.0000 |
| M1105 | uVSG | - | … | 0.9614 | 0.0000 |
| M1287 | kVSG | NC_007924_Lactobacillus_phage_KC5a | … | 0.9999 | 0.0000 |
| M285 | uVSG | - | … | 0.9813 | 0.0000 |
| M349 | kVSG | NC_029012_Parabacteroides_phage_YZ2015a | … | 0.9998 | 0.0000 |
| M616 | uVSG | - | … | 0.0000 | 0.9975 |
| M651 | uVSG | - | … | 1.0000 | 0.0000 |
| M697 | uVSG | - | … | 0.0000 | 0.9941 |
| M738 | uVSG | - | … | 0.0000 | 1.0000 |
You can refer to these tables: VSG-to-species and VSG-to-SGBs CSV files to check the annotations of each group. For example, the top 5 hits for group M1102 are:
| Group | Group-Type [k|u] | … | Species | SGB | hits | genomes in the bin |
|---|---|---|---|---|---|---|
| M1102 | kVSG | … | Parabacteroides_merdae | kSGB1949 | 34 | 2296 |
| M1102 | kVSG | … | Phocaeicola_dorei | kSGB1815 | 10 | 1473 |
| M1102 | kVSG | … | Campylobacter_coli | kSGB19443 | 9 | 6192 |
| M1102 | kVSG | … | Parabacteroides_distasonis | kSGB1934 | 7 | 4705 |
| M1102 | kVSG | … | Bacilli_bacterium | kSGB1814 | 4 | 1335 |
| M1102 | kVSG | … | ... | ... | ... | ... |
Where hits indicates the number of genomes of that species / SGB with a match against a member of that viral group.
(in a future version, this will be integrated in the standard output)