melonnpan
MelonnPan (Model-based Genomically Informed High-dimensional Predictor of Microbial Community Metabolic Profiles) is a computational method for predicting metabolite composition from microbiome sequencing data.
Support is available from the MelonnPan bioBakery Forum.
For additional information, see the MelonnPan User Manual.
Citation:
Himel Mallick, Eric A. Franzosa, Lauren J. McIver, Soumya Banerjee, Alexandra Sirota-Madi, Aleksandar D. Kostic, Clary B. Clish, Hera Vlamakis, Ramnik Xavier, Curtis Huttenhower (2019). Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nature Communications 10(1):3136-3146.
Table of contents
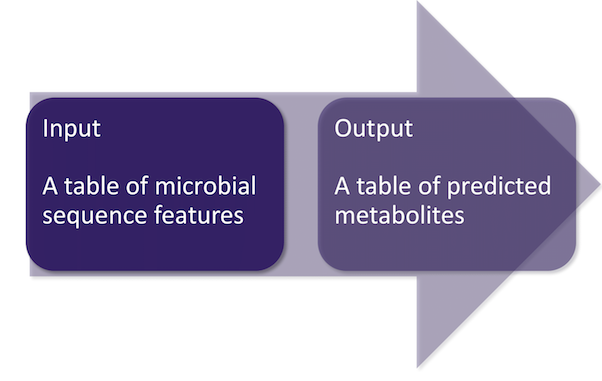
- R software ( version >= 3.5.0)
Install MelonnPan by first obtaining the source code. Then install the dependencies and install the software. Run the following commands to install MelonnPan and its dependencies:
$ git clone https://github.com/biobakery/melonnpan.git
$ R -q -e "install.packages(c('devtools', 'glmnet', 'foreach', 'getopt', 'doParallel', 'vegan', 'data.table', 'ggplot2', 'AssocTests', 'optparse', 'tibble'), repos='http://cran.r-project.org')"
$ R CMD INSTALL melonnpan
This section presents an example run of MelonnPan-Predict.
The default MelonnPan-Predict function uses a pre-trained model from the human gut based on UniRef90 gene families (functionally profiled by HUMAnN2). Download the example test data melonnpan.test.data.txt to get started on the tutorial. This dataset contains functional profiles (i.e. quality-controlled UniRef90 gene families ) from 65 IBD and non-IBD subjects from the NLIBD cohort (as described in Franzosa et al., 2019 and Mallick et al., 2019). These functional profiles were generated using HUMAnN2 and normalized to proportions. Gene family abundances were passed through basic filtering to remove low-variance features.
If you open up this file in a spreadsheet editor like Excel, you should see:
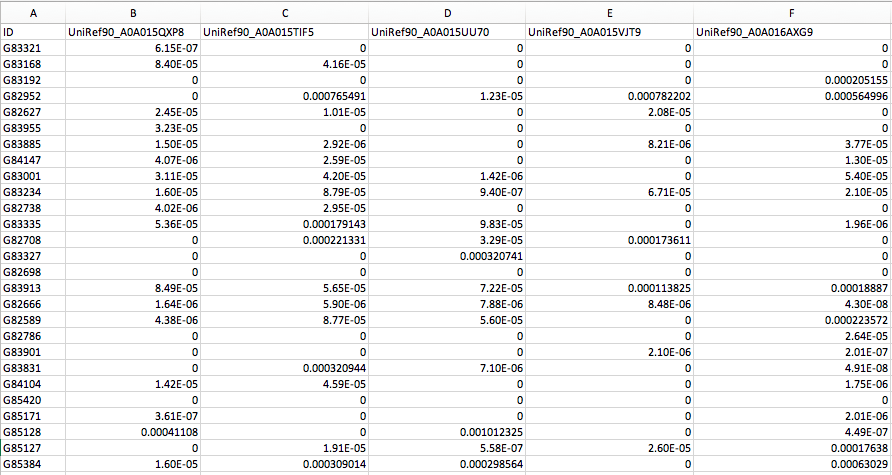
Assuming that the downloaded file is saved in the Downloads directory,
let’s run MelonnPan-Predict on this demo input file, pplacing the output
files in the Output folder within your current working directory:
$ cd ~/Downloads
$ Rscript predict_metabolites.R -i melonnpan.test.data.txt -o Output/
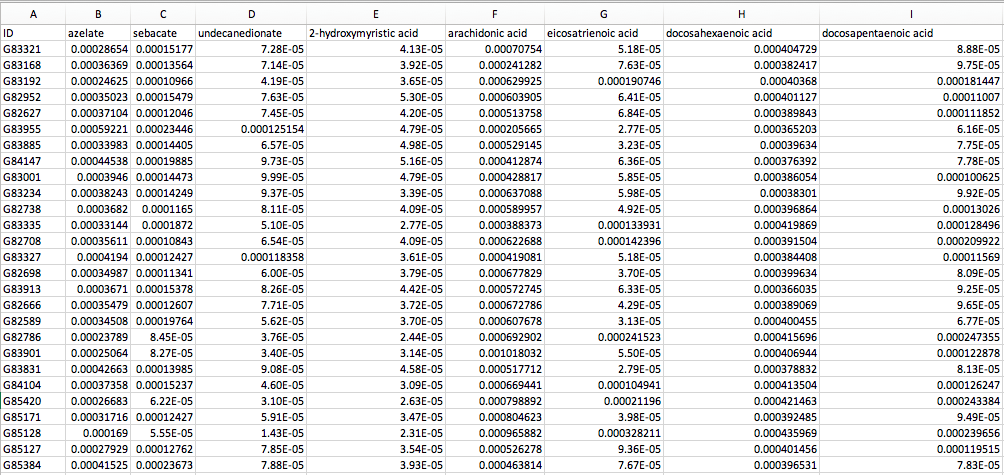
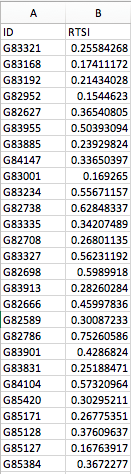
The command above creates two primary output files:
- MelonnPan_Predicted_Metabolites.txt
- MelonnPan_RTSI.txt
The first file contains the predicted metabolite relative abundances and the second file records RTSI scores per sample.
MelonnPan_Predicted_Metabolites.txt
MelonnPan_RTSI.txt
This section presents an example run of MelonnPan-Train.
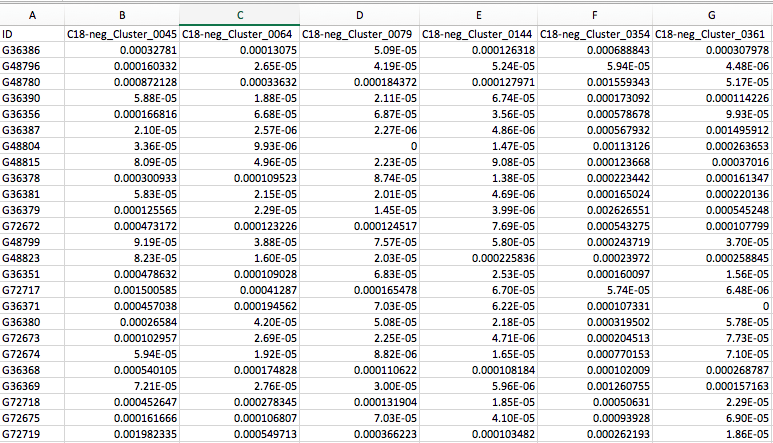
The default MelonnPan-Train function uses an elastic net regularization to train metabolites using microbial sequence features. For the purpose of this tutorial, we will use a version of the PRISM metabolites (as described in Franzosa et al., 2019 and Mallick et al., 2019) melonnpan.training.compounds.txt. This dataset contains metabolite compounds from 157 IBD and non-IBD subjects in the PRISM cohort generated using LC-MS profiling. As above, metabolite abundances were passed through basic filtering to remove low-variance features.
If you open up this file in a spreadsheet editor like Excel, you should see the following:
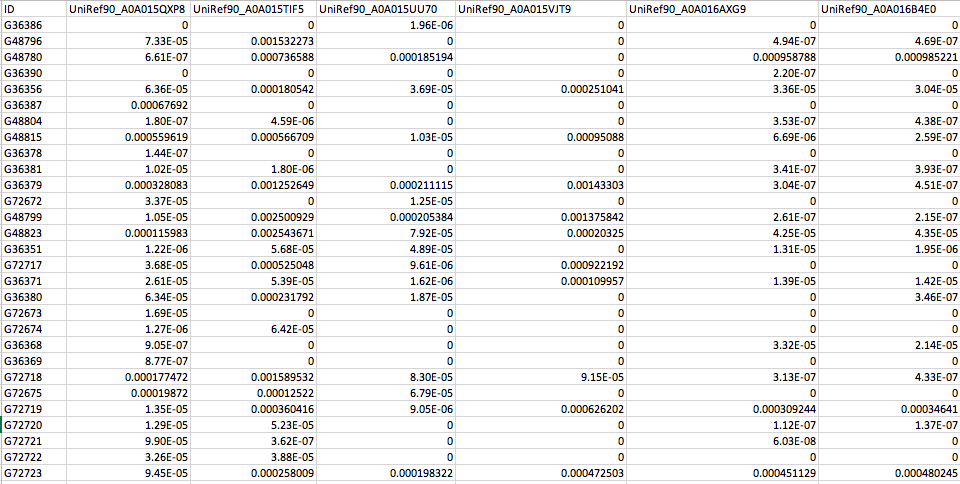
We also need to supply the corresponding metagenomes for the same subjects. Here we use a preprocessed table of UniRef90 gene families from the same cohort, functionally profiled by HUMAnN2 (similar to the MelonnPan-Predict example above), which looks like the following:
NOTE: The above two input files contain values normalized to relative abundances and they have the matching sample names as the first column.
Assuming that the downloaded files are saved in the Downloads directory,
let’s run MelonnPan-Train on these demo input files, placing the output
files in the Output folder within your current working directory:
$ cd ~/Downloads
$ Rscript train_metabolites.R -i melonnpan.training.compounds.txt -g melonnpan.training.data.txt -o Output/
The command above creates three primary output files (note that this step is computationally intensive and we recommend running with multiple cores for best results):
- MelonnPan_Trained_Metabolites.txt
- MelonnPan_Trained_Weights.txt
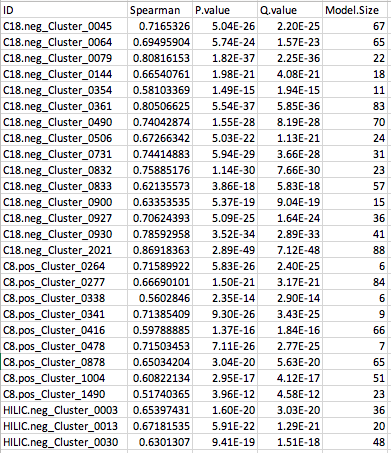
- MelonnPan_Training_Summary.txt
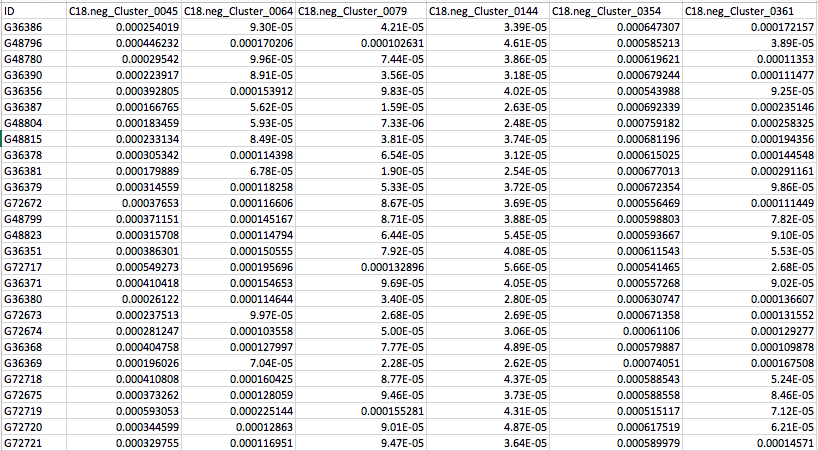
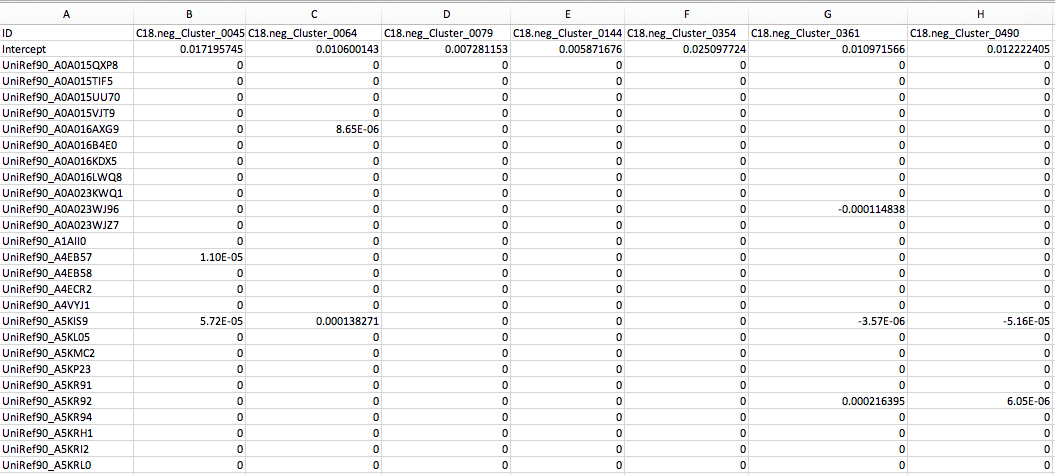
The first file contains the predicted metabolite relative abundances and the second and third file record training weights and summary respectively.
MelonnPan_Trained_Metabolites.txt
MelonnPan_Trained_Weights.txt
MelonnPan_Training_Summary.txt
NOTE: MelonnPan-Train is reference-agnostic, which means, it can be run for any combination of MS-based metabolomics data (LC-MS, GC-MS, NMR, etc.) and sequence features' data (amplicon or metagenomic taxonomic or functional profiles) provided they are measured from the same biological specimens (i.e. the datasets are paired).
This tutorial is still in development but we will periodically update the tutorial to include other use of MelonnPan. Some specific next steps include:
- Visualization of prediction results
- Customizing MelonnPan-Train for arbitrary predictors (i.e. different from the default UniRef90)
- Customizing MelonnPan-Predict for arbitrary weight matrix (i.e. different from the default weight)
Check back soon for more updates!