shortbred
ShortBRED (Short, Better Representative Extract Dataset) is a pipeline to take a set of protein sequences, cluster them into families, build consensus sequences to represent the families, and then reduce these consensus sequences to a set of unique identifying strings ("markers"). The pipeline then searches for these markers in metagenomic data and determines the presence and abundance of the protein families of interest.
For additional information, please refer to the manuscript: Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, Huttenhower C.High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput Biol. 2015 Dec 18;11(12):e1004557.
We provide support for ShortBRED users. Feel free to post any questions on the bioBakery forum.
Table of contents
The following figure shows the workflow of ShortBRED (Fig. 1 in the manuscript).
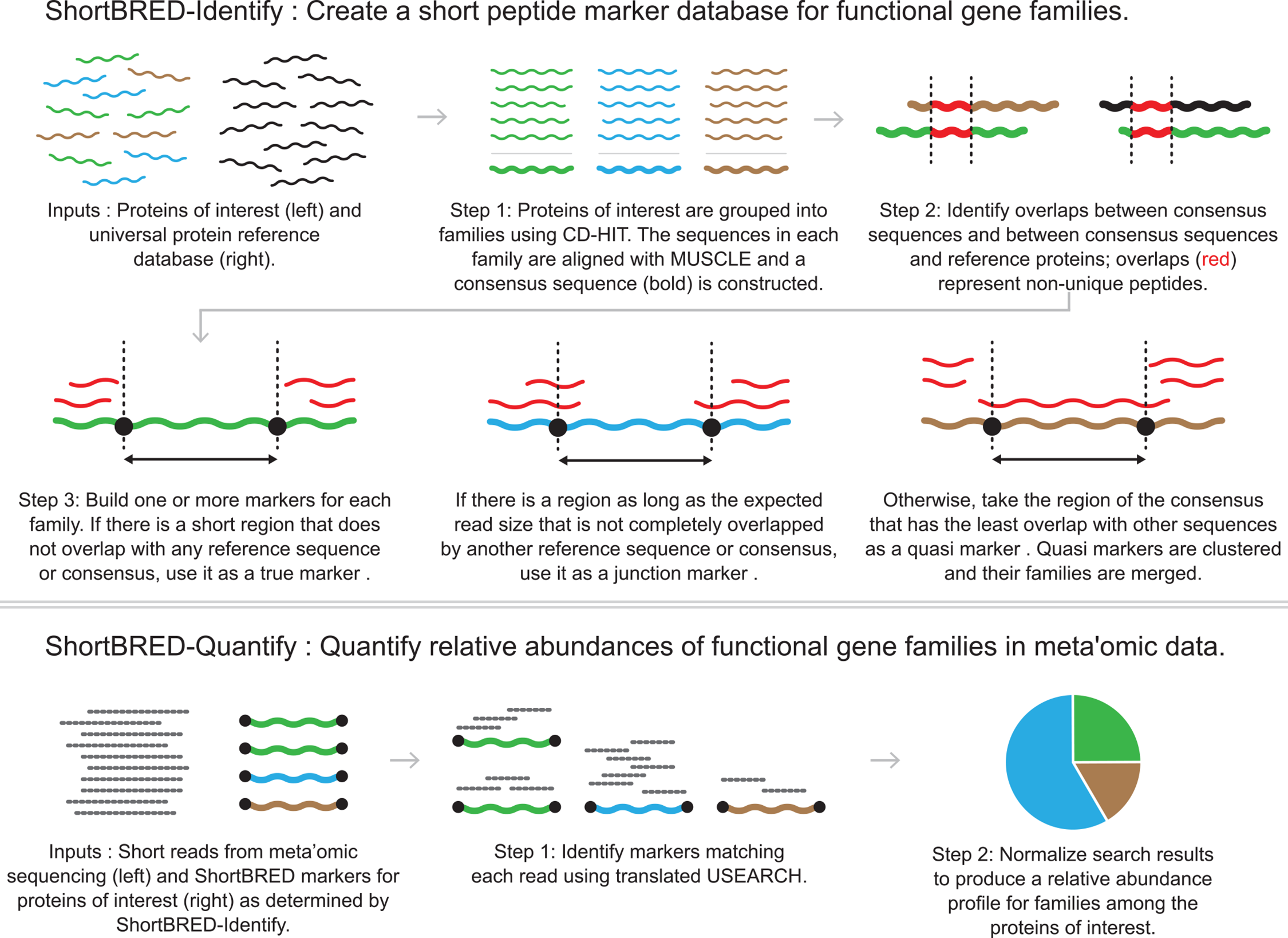
ShortBRED can be installed with Conda or run from a Docker image. Please note, if you are using bioBakery (Vagrant VM or cloud) you do not need to install ShortBRED because the tool and its dependencies are already installed. However, you will need to install the dependency USEARCH which requires a license. Follow the commands in the instructions to install bioBakery dependencies that require licences.
Install with Conda: $ conda install -c biobakery shortbred Note: Before
installing with conda update your channels so biobakery is at the top of
the list.
Install with Docker: $ docker run -it biobakery/shortbred bash
If you would like to install from source, refer to the ShortBRED user manual for the pre-requisites/dependencies and installation instructions.
The input files required for ShortBRED are the following:
- Proteins of interest - amino acid sequences of the proteins you are looking for.
- Reference proteins - a collection of 'background' protein sequences to help rule out non-unique regions of the proteins of interest; this should be as exhaustive as possible, e.g. UniRef90
For this tutorial, our proteins of interest are a small set of TonB genes from Proteobacteria. TonB is a siderophore import protein found in the outer membrane of gram negative bacteria, and the reference proteins are subsampled from the integrated microbial genomes (IMG) database. Both are located in the example directory under the main ShortBRED page
Let's examine these protein sequences.
$ less tonB1. Are there obvious regions of overlap between the TonB genes?
2. Is there overlap between TonB and the reference proteins?
We're trying to identify tonB genes in set of short DNA reads from metagenomic sequencing. The data we'll use for the tutorial was synthetically generated to contain 1,000 reads from tonB genes, and 10,000 reads from other proteins.
To show why ShortBRED is useful, we will start by doing a naive BLAST search. First, make a BLAST database combining our tonB genes, and our reference database:
$ cat tonB.faa ref_prots.faa > allprots.faa
$ makeblastdb -dbtype prot -in allprots.faa -out allprots
Building a new DB
New DB name: allprots
New DB title: allprots.faa
Sequence type: Protein
Keep Linkouts: T
Keep MBits: T
Maximum file size: 1000000000B
Adding sequences from FASTA; added 83 sequences in 0.00298595 seconds.Now, we'll BLAST our DNA reads against this protein database using translated search of the short reads against the sequences of our proteins of interest and the reference sequences. The following command searches the database for alignments to our reads and generates a table with the query (read) ID, the subject (database) ID, the percent identity and the length of the alignment:
$ blastx -query shortbred_demo_reads.fasta -db allprots \
-out naive_search.tsv -outfmt "6 qseqid sseqid pident length"First, notice how long this takes. Our database is tiny, and we don't have many reads, but translated search is SLOW.
Why is translated search slow compared with nucleotide search?
Let's take a look at the results (once you run this command, press <space> to scroll through, and q to exit back to the command prompt):
$ column -t naive_search.tsv | less
Background_read|00000001|0062|-1|WP_037108668.1 tonB_ACD08273 84.375 32
Background_read|00000001|0062|-1|WP_037108668.1 tonB_ADB98050 84.375 32
Background_read|00000001|0062|-1|WP_037108668.1 tonB_EGT66731 84.375 32
Background_read|00000001|0062|-1|WP_037108668.1 tonB_SAP90706 84.375 32
Background_read|00000001|0062|-1|WP_037108668.1 tonB_CDO13422 87.500 32
Background_read|00000001|0062|-1|WP_037108668.1 tonB_AIE04533 84.375 32
Background_read|00000001|0062|-1|WP_037108668.1 ref_637982072 45.000 20
Background_read|00000001|0062|-1|WP_037108668.1 ref_637982015 40.000 10
Background_read|00000001|0062|-1|WP_037108668.1 ref_637982013 58.333 12
Background_read|00000002|0003|-1|WP_037108668.1 ref_637982016 70.000 10
Background_read|00000002|0003|-1|WP_037108668.1 ref_637982016 50.000 14
Background_read|00000002|0003|-1|WP_037108668.1 ref_637982011 37.500 16Let's check how many hits came from actual TonB genes vs background (searching here for hits of at least 85% identity and at least 25aa long). The following command searches through the results file looking for lines that have TonB a the beginning (grep -E '^TonB'), tonB_ somewhere else (grep -E 'tonB_'), and where column 3 has a value > 85, and column 4 has a value > 25 (awk '$3 > 85 && $4 > 25 {print $0}'). Finally, it returns a count of the lines (wc is "word count", and the -l flag asks it to count lines).
$ cat naive_search.tsv | grep -E '^TonB' | grep -E 'tonB_' | awk '$3 > 85 && $4 > 25 {print $0}' | wc -l
3153
$ cat naive_search.tsv | grep -E '^Background' | grep -E 'tonB_' | awk '$3 > 85 && $4 > 25 {print $0}' | wc -l
15941. What's the maximum length hit we'd expect, if our reads are 100bp long?
2. Why do we get more than 3,000 hits for tonB, when we know that there were only 1000 matching reads in our metagenome?
Transition discussion: How do you create your database of proteins of interest?
Now let’s run ShortBRED. ShortBRED-Identify clusters the input protein sequences into families, builds consensus sequences, and then identifies regions of overlap among the consensus sequences and between the consensus sequences and a set of reference proteins. This information is used to construct a set of representative markers for the families.
Why might proteins share regions of overlap even if they are not directly related (homologous)?
ShortBRED begins with the same pair of files, which can be found in the input/ folder:
First, run the shortbred_identify.py -h to see the help page for this step.
To create markers for the sample data, run the following command from the ShortBRED working directory:
$ shortbred_identify.py --goi tonB.faa --ref ref_prots.faa --markers mymarkers.faa --tmp example_identify
Checking dependencies...
Tested usearch. Appears to be working.
Tested blastp. Appears to be working.
Tested muscle returned a nonzero exit code (typically indicates failure). Please check to ensure the program is working. Will continue running.
Path for cdhit appears to be fine. This program returns an error [exit code=1] when tested and working properly, so ShortBRED does not check it.
Tested makeblastdb. Appears to be working.
Checking to make sure that installed version of usearch can make databases...
Usearch appears to be working.
Clustering proteins of interest...
================================================================
Program: CD-HIT, V4.7 (+OpenMP), Jul 11 2017, 08:30:58
Command: cd-hit -i tonB.faa -o
example_identify/clust/clust.faa -d 0 -c 0.85 -b 10 -g
1The above command will create a set of markers for TonB proteins (mymarkers.faa). An example of the output is below:
$ head -20 mymarkers.faa
>tonB_AIE04533_TM_#01
MRLPAFYRWLLLVVGISISGISLAQDAGWPRQIQDSRGVHTLDHKPA
>tonB_AIE04533_TM_#02
DVAKARHVARLYIGEPNAETVAAQMPDLILISATGGDSALALYDQLSAIAPTLVINYDDK
SWQSLLTQLGEITGQEKQAAARIAEFETQLTTVKQRIALPPQPVSALVYTPAAHSANLWT
PESAQGKLLT
>tonB_AIE04533_TM_#03
TLLLNRLAALF
>tonB_CDO13422_TM_#01
MNFFSFCRRGALTGMLLLLGITSA
>tonB_CDO13422_TM_#02
YGTHTLPSQPL
>tonB_CDO13422_TM_#03
EVAKARKLA
>tonB_CDO13422_TM_#04
KTIAPTLVINYDDKSWQTLLTQLGQITGHEQQASARIADFNKQLVSLKEKM
>tonB_CDO13422_TM_#05
LLVLQRLSSLFG
>tonB_ABS46221_TM_#01
MNKTRTCTTLAPRWLGDISFILAGLLALFFLLSVGDSQAETGAVHTTQANGFPRK
What do the first three sequences of the markers file represent?
The directory example_identify (folder name provided with the tmp flag) should contain the processed data from ShortBRED (including blastresults). Please refer to the documentation for further details.
ShortBRED-Quantify then searches for the markers in short-read sequencing data, and returns a normalized, relative abundance table of the protein families found. This script takes the FASTA file of markers and quantifies their relative abundance in a FASTA file of nucleotide metagenomic reads.
The input files required for the script are the following (Sample input files for the purpose of this tutorial are provided) :
- Markers file (generated from Section 3)
- Short nucleotide reads - shortbred_demo_reads.fasta
ShortBRED uses the markers file to generate a blast database for our short reads, focusing specifically on the unique markers. This makes the search substantially faster and more specific.
First, run the shortbred_quantify.py -h to see the help page for this step.
To create markers for the sample data, run the following command from the shortbred working directory: :
$ shortbred_quantify.py --markers mymarkers.faa --wgs shortbred_demo_reads.fasta --results shortbred_results.tsv --tmp example_quantifyThe above command will create an output file shortbred_results.tsv containing normalized count data of the protein families in the wgs data. An example of the output is below:
$ column -t shortbred_results.tsv
Family Count Hits TotMarkerLength
tonB_ABS46221 12269.938650306749 76 300
tonB_AIE04533 8741.258741258742 58 188
tonB_CAE16995 15289.25619834711 75 279
tonB_CDO13422 5509.641873278237 23 107
tonB_EGT66731 37878.78787878788 92 102
Column descriptions:
-
Family - The family/cluster to which the marker belongs.
-
Count - This is the normalized count for the marker described in the section “Profiling protein family metagenomic abundance with ShortBRED-Quantify” of the ShortBRED manuscript. It is expressed in units of RPKMs (reads per kilobase of reference sequence per million sample reads). Please note that the “final” result ShortBRED calculates is at the family level. It takes the median of these marker normalized counts as the family level value.
-
Hits - The number of valid ShortBRED hits to the marker.
-
TotMarkerLength - The length of the marker in amino acids.
1. We know there were 1000 reads mapping to TonB proteins. How many of them were identified?
2. What is the false positive rate of ShortBRED in this case?
ShortBRED uses BLAST under the hood, and you can explore the results of the search in the same way as we did above:
$ cat example_quantify/wgsfull_results.tab | grep -E '^TonB' | grep -E 'tonB_' | awk '$3 > 85 && $4 > 25 {print $0}' | wc -l
276
$ cat example_quantify/wgsfull_results.tab | grep -E '^Background' | grep -E 'tonB_' | awk '$3 > 85 && $4 > 25 {print $0}' | wc -l
15The directory example_quantify (folder name provided with the tmp flag) should contain the processed data from ShortBRED. Please refer to the documentation for further details.
1. ShortBRED favors specificity (avoiding false positives) over perfect sensitivity (detecting all true positives). Under what circumstances would this be important?
2. Under what circumstances would it be inadvisable?
3. What is a scenario where ShortBRED will have a false negative where the naive BLAST search would not?
Discussion : Under what conditions would you choose to use shortBRED over HUMAnN?