Whole Genome Sequencing Clinical Assay
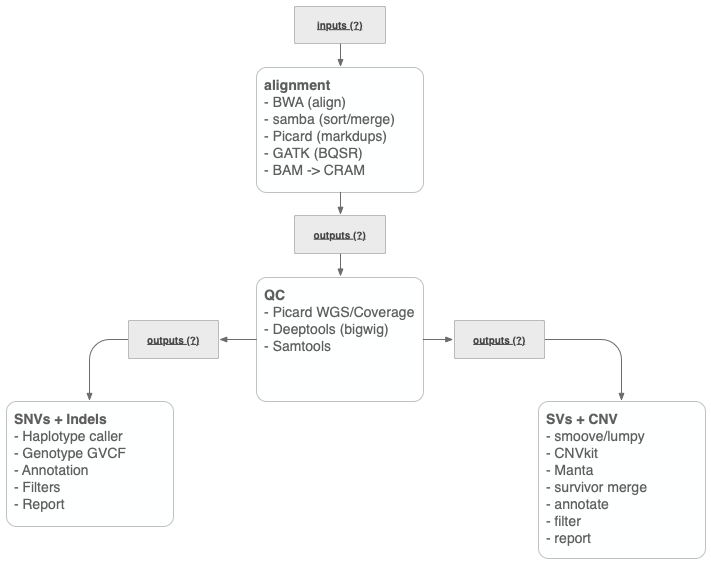
The Whole Genome Sequencing (WGS) Clinical Assay combines the DNA Alignment, WGS Quality Control, Germline SNVs and small Indels, and Single Sample SVs subworkflows into one WGS workflow. Please see each linked Wiki above for additional details regarding each subworkflow.
Please see each subworkflow listed above for a command line tutorial that exemplifies each step that is run as part of the entire workflow.
The Common Workflow Language (CWL) definitions for the entire workflow are contained in the Analysis Workflows GitHub repository. Specifically, the germline_wgs.cwl is the pipeline definition that will run the entire process including each subworkflow listed in the Introduction.
| Name | Description | Example | Subworkflow |
|---|---|---|---|
| bams | DNA Alignment | ||
| bqsr_intervals | DNA Alignment | ||
| dbsnp_vcf | DNA Alignment | ||
| known_indels | DNA Alignment | ||
| mills | DNA Alignment | ||
| readgroups | DNA Alignment | ||
| reference | DNA Alignment, WGS Quality Control, Germline SNVs and small Indels, Single Sample SVs | ||
| annotate_coding_only | Germline SNVs and small Indels | ||
| emit_reference_confidence | GVCF | Germline SNVs and small Indels | |
| gvcf_gq_bands | NULL | Germline SNVs and small Indels | |
| intervals | chr1 .. chr22, chrX, chrY | Germline SNVs and small Indels | |
| synonyms_file | Germline SNVs and small Indels | ||
| variant_reporting_intervals | Germline SNVs and small Indels | ||
| vep_cache_dir | Location of a local ensembl cache to be used by vep | Germline SNVs and small Indels, Single Sample SVs | |
| vep_ensembl_assembly | Which species assembly version vep should use | GRCh38 | Germline SNVs and small Indels, Single Sample SVs |
| vep_ensembl_species | Which species vep should use | homo_sapiens | Germline SNVs and small Indels, Single Sample SVs |
| vep_ensembl_version | Which ensembl release vep should use | 95 | Germline SNVs and small Indels, Single Sample SVs |
| cnvkit_diagram | Create an ideogram of copy ratios on chromosomes as a pdf | FALSE | Single Sample SVs |
| cnvkit_drop_low_coverage | Helps avoid false positive deletions in low quality tumor samples | FALSE | Single Sample SVs |
| cnvkit_male_reference | Use/assume a male reference | FALSE | Single Sample SVs |
| cnvkit_method | Sequencing protocol used | wgs | Single Sample SVs |
| cnvkit_reference_cnn | A copy number reference file against which potential copy number variants will be evaluated | Single Sample SVs | |
| cnvkit_scatter_plot | Create a whole genome copy ratio profile as a pdf scatter plot | Single Sample SVs | |
| cnvkit_vcf_name | custom name to use for the cnvkit output vcf | Single Sample SVs | |
| manta_call_regions | Single Sample SVs | ||
| manta_non_wgs | Single Sample SVs | ||
| manta_output_contigs | Single Sample SVs | ||
| maximum_sv_pop_freq | Single Sample SVs | ||
| merge_estimate_sv_distance | TRUE | Single Sample SVs | |
| merge_max_distance | 1000 | Single Sample SVs | |
| merge_min_sv_size | 1 | Single Sample SVs | |
| merge_min_svs | Single Sample SVs | ||
| merge_same_strand | TRUE | Single Sample SVs | |
| merge_same_type | TRUE | Single Sample SVs | |
| merge_sv_pop_freq_db | Single Sample SVs | ||
| smoove_exclude_regions | Single Sample SVs | ||
| sv_filter_interval_lists | Single Sample SVs | ||
| sv_variants_to_table_fields | CHROM, POS, ID, REF, ALT, SVLEN, CHR2, END, POPFREQ_AF, POPFREQ_VarID, NSAMP | Single Sample SVs | |
| sv_variants_to_table_genotype_fields | GT | Single Sample SVs | |
| custom_clinvar_vcf | WGS Quality Control | ||
| custom_gnomad_vcf | WGS Quality Control | ||
| minimum_base_quality | WGS Quality Control | ||
| minimum_mapping_quality | WGS Quality Control | ||
| omni_vcf | WGS Quality Control | ||
| per_base_intervals | WGS Quality Control | ||
| per_target_intervals | WGS Quality Control | ||
| picard_metric_accumulation_level | WGS Quality Control | ||
| qc_intervals | WGS Quality Control | ||
| summary_intervals | WGS Quality Control |
| Filename | Pipeline Step | Software | Description |
|---|---|---|---|
| annotated.coding_variant_filtered.vcf.gz | |||
| annotated.coding_variant_filtered.vcf.gz.tbi | |||
| annotated.max_sv_pf_filtered.vcf | |||
| annotated.vcf.gz | |||
| annotated.vcf.gz.tbi | |||
| annotated.vcf_summary.html | |||
| candidateSmallIndels.vcf.gz | |||
| candidateSmallIndels.vcf.gz.tbi | |||
| candidateSV.vcf.gz | |||
| candidateSV.vcf.gz.tbi | |||
| chr##.g.vcf.gz | |||
| chr##.g.vcf.gz.tbi | |||
| final.AlignmentSummaryMetrics.txt | |||
| final.antitargetcoverage.cnn | |||
| final.bam.bigwig | |||
| final.bam.flagstat | |||
| final.bam.merged.NameSorted.mark_dups_metrics.txt | |||
| final.base-clinvar-HsMetrics.txt | |||
| final.base-clinvar-PerBaseCoverage.txt | |||
| final.cnr | |||
| final.cns | |||
| final.cnvkit.vcf | |||
| final.crai | |||
| final.cram | |||
| final.cram.crai | |||
| final.InsertSizeHistogram.pdf | |||
| final.InsertSizeMetrics.txt | |||
| final.target-acmg_genes-HsMetrics.txt | |||
| final.target-acmg_genes-PerTargetCoverage.txt | |||
| final.targetcoverage.cnn | |||
| final.target-gencode_exons-HsMetrics.txt | |||
| final.target-gencode_exons-PerTargetCoverage.txt | |||
| final.target-gencode_genes-HsMetrics.txt | |||
| final.target-gencode_genes-PerTargetCoverage.txt | |||
| final.VerifyBamId.depthSM | |||
| final.VerifyBamId.selfSM | |||
| GcBiasMetricsChart.pdf | |||
| GcBiasMetricsSummary.txt | |||
| GcBiasMetrics.txt | Alignment and QC | CollectGcBiasMetrics (Picard) | See https://broadinstitute.github.io/picard/picard-metric-definitions.html#GcBiasMetrics |
| intersect-acmg_genes.vcf.gz | |||
| intersect-acmg_genes.vcf.gz.tbi | |||
| intersect-gencode_exons.vcf.gz | |||
| intersect-gencode_exons.vcf.gz.tbi | |||
| intersect-gencode_genes.vcf.gz | |||
| intersect-gencode_genes.vcf.gz.tbi | |||
| select_variants.vcf.gz | |||
| select_variants.vcf.gz.tbi | |||
| sorted.vcf | |||
| SV-smoove.genotyped.vcf.gz | |||
| SV-smoove.genotyped.vcf.gz.csi | |||
| tumorSV.vcf.gz | |||
| tumorSV.vcf.gz.tbi | |||
| variants.annotated.tsv | |||
| WgsMetrics.txt |